
Abstract
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by an abnormal expansion of CAG repeats. Although pathogenesis has been attributed to this polyglutamine expansion, the underlying mechanisms through which the huntingtin protein functions have yet to be elucidated. It has been suggested that postnatal reduction of mutant huntingtin through protein interference or conditional gene knockout could prove to be an effective therapy for patients suffering from HD. For allele-specific targeting, transcription activator-like effectors (TALE) were designed to target single-nucleotide polymorphisms (SNP) in the mutant allele and packaged into a vector backbone containing KRAB to promote transcriptional repression of the disease-associated allele. Additional TALEs were packaged into a vector backbone containing heterodimeric FokI and were designed to be used as nucleases (TALEN) to cause a CAG-collapse in the mutant allele. Human HD fibroblasts were treated with each TALE-SNP or TALEN. Allele-expression was measured using a SNP-genotyping assay and mutant protein aggregation was quantified with Western blots for anti-ubiquitin. The TALE-SNP and TALEN significantly reduced mutant allele expression (p < 0.05) when compared to control transfections while not affecting expression of the nondisease allele. This study demonstrates the potential of allele-specific gene modification using TALE proteins, and provides a foundation for targeted treatment for individuals suffering from Huntington's or other genetically linked diseases.
Keywords
Introduction
Huntington's disease (HD) is an inherited autosomal-dominant neurodegenerative disease caused by an expansion of CAG trinucleotide repeats found in exon 1 of the huntingtin (HTT) gene (43). Individuals with 38 or more CAG repeats typically develop HD in adulthood (12), and greater than 60 CAG repeats results in in juvenile (JHD) onset (34). The expanded CAG region causes the production of mutant huntingtin protein and results in progressive degeneration of neurons, primarily in the putamen, caudate nucleus, and cerebral cortex (1,37). HD is clinically diagnosed by perturbed motor functioning termed chorea, or involuntary muscle movements. Neurological symptoms of HD include cognitive impairment affecting processes such as reasoning, memory, and comprehension. Psychologically, HD patients manifest personality changes along with depression, anxiety, and other emotional disturbances (25,39). Behavioral abnormalities including dementia and impaired motor and speech functions occur in a progressive fashion (24,33), ultimately rendering patients incapable of caring for themselves. HD eventually culminates in death around 15–20 years after the diagnoses of motor symptoms. Despite the wealth of clinical research accumulated since the identification of the mutant Huntington gene published in 1993 (43), there continues to be no cure for HD; only palliative therapies aimed at reducing motor symptoms (21,23) and psychological disturbances are available.
Although HD has a single genetic cause, the disease pathology is very complex, leading to detrimental effects on a wide variety of cellular processes. In addition to the complexity of the disorder, our lack of knowledge of the exact function of the healthy and mutant protein makes it difficult to develop an appropriate therapy (13,19). Healthy huntingtin is essential for neuronal development (29,32), and new studies have pointed toward the importance of the protein in adulthood (35). Mutant huntingtin accumulates in the cells of HD patients, aggregating in the nucleus and causing alterations in gene transcription that result in toxicity (2,4,39). This aggregation results in downstream deficits such as dysfunction in RNA synthesis, the ubiquitin proteasome system, impaired mitochondrial activity, cellular inclusions, and activation of proapoptotic molecules that eventually lead to cell death (5,14,17). Due to the development of transgenic mouse models of HD, studies have begun to uncover the widespread pathological effects of mutant huntingtin. Conditional knockout of mutant huntingtin in the striatum has demonstrated partial motor and psychiatric recovery, but to ameliorate HD-like symptoms, striatal and cortical knockout of mutant huntingtin was required (46). Additionally, studies have shown widespread HD-associated pathology in the brain and other organs of transgenic mice (6,30,31,4). Any potential avenue for HD therapy must take into account the role of the healthy huntingtin protein in maintaining neuronal health, as complete suppression of both wild-type and mutant alleles has resulted in unintended negative consequences (8,35,44).
There have been several encouraging studies suggesting that postnatal reduction of mutant huntingtin through protein interference or conditional gene knockout may be an effective therapy for patients suffering from HD (15,22,26,36,41,42). Allele-specific silencing of the huntingtin gene may be possible by either targeting the expanded CAG region directly or by targeting genetic polymorphisms found in the mutant allele. Despite the genetic diversity around the huntingtin gene, 190 single nucleotide polymorphisms (SNP) that are associated with the mutant allele have been identified in adult patients (47). The minor allele frequency (MAF) for a large portion of the validated SNP was greater than 0.20, meaning that these targetable polymorphisms occur in at least 20% of the population. The strong association between specific SNP alleles and CAG expansion also provides an opportunity for personalized therapeutics in HD in which the clinical development of only a small number of allele-specific targets may be sufficient to treat up to 88% of the HD patient population (47).
We targeted the mutant huntingtin allele through two approaches: transcriptional repression and gene correction. To achieve the allele-specific transcriptional repression of mutant HTT, we designed transcription activator-like effectors (TALE) to target unique DNA sequences in the mutant allele (9,10,27). These TALEs can be paired with a variety of effector domains to modulate gene expression or to have specific nuclease activity (20,49). For the gene silencing approach, we constructed TALEs targeting SNPs that occur preferentially in the mutant Huntingtin allele, and thus serve to distinguish the mutant allele from the healthy allele. We selected three SNP sites that occur independently of each other in greater than 45% of the HD population to target with our gene repression strategy. The targeted SNPs used in this study were rs762855 (MAF 0.45; mutant allele association 0.97; referred to as T7 in the figures), rs3856973 (MAF 0.48; mutant allele association 0.94; referred to as T3γ in the figures), and rs2024115 (MAF 0.48; mutant allele association 0.97; referred to as T2 in the figures). These TALEs are paired with a transcriptional repressor Krüppel associated box (KRAB) to silence gene expression upon binding (28,40,48).
For the gene correction approach, pairs of TALEs were attached to obligate-heterodimeric variants of the nuclease from Flavobacterium okeanokoites (FokI) (TALEN). Our rationale for the use of a heterodimeric nuclease was to reduce potential off-target nuclease activity by necessitating complementary binding of the two TALE arrays to elicit a double-stranded break. The breaks were expected to be repaired by the highly efficient single-strand annealing pathway, resulting in the deletion of CAG repeats (7,11). The TALEN pairs targeted the forward and reverse strands of the CAG repeats (referred to as CAG-F&R in the figures). This design takes into account the number of repeats required for each TALEN to bind and the space needed for FokI to create a double-stranded break, such that only CAG lengths greater than 15 would enable TALEN-mediated double-strand breaks. Therefore, only long (pathogenic) CAG repeats would be substrates for CAG shortening.
Our data demonstrate that these two gene-targeting approaches have the potential to become potent therapies through allele-specific reduction of the mutant huntingtin allele. In addition, this technology can easily be adapted to a wide-range of trinucleotide repeat disorders (38) and other diseases for which there is a known genetic mutation, thus expanding the utility of this approach and making it a highly favorable therapeutic strategy. Here, we explore the efficacy of allele-specific targeting of the mutant hungtingtin allele in patient-derived fibroblasts.
Materials and Methods
TALE Construction
TALEs were constructed using the Golden Gate TALEN kit (Addgene, Cambridge MA, USA) following previous published protocols (9). All TALEs were initially cloned into pTAL2 vector (Addgene) before subsequent insertion into a VP64-fused vector for luciferase confirmation assay. TALEs were further cloned into a phosphoglycerate kinase plasmid (pPGK) vector with KRAB-fused, FokI DD, or RR effector domains (3) (Addgene) for gene silencing or CAG collapse, respectively. Each TALE was designed for a unique 18 base pair sequence only found in either the promoter region of the huntingtin gene or the CAG expansion. For transcriptional repression of the mutant allele the TALE was designed to have the HD-associated SNP occurring in the first four repeat variable diresidues of the TALE plasmid.
Luciferase Binding Assay
The luciferase binding assay is a conformational assay developed to measure the binding and biological activity of the TALE. Briefly, the target sequence for the TALE is cloned into the promoter of a luciferase reporter gene and transfected into HEK293T cells (ATCC, Manassas, VA, USA). Upon binding to the correct location the TALE should turn on the reporter gene and the cells will emit luciferase demonstrating the binding of the TALE. TALE binding sites were inserted in front of the SV40 promoter in a pGL3 Luciferase Reporter vector (3) (Addgene) in between the NheI and XhoI sites via PCR for each TALE constructed. VP64-fused TALEs were cotransfected with the pGL3 Luciferase Reporter into HEK293T cells using Lipofectamine 3000 (Life Technologies, Carlsbad, CA, USA). Luciferase assays were performed 2 days posttransfection in a Veritas Microplate Luminometer using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) protocol.
HD Fibroblast Culture and Transfections
To perform the initial proof-of-concept studies of allelespecific correction or silencing, we wanted to test the therapeutic in an easy-to-isolate and -expand cell line that contained the naturally occurring gene mutation under the endogenous promoter. Human HD fibroblasts were purchased from Coriell Cell Repository (GM02151 Female 26 year old; Camden, NJ, USA) and cultured at 37°C, 5% CO2, in DMEM high glucose (Hyclone, Logan, UT, USA) supplemented with 20% Premium Select FBS (Atlanta Biologicals, Norcross, GA, USA) and 1% L-glutamine (Hyclone, Logan, UT, USA). The study was IRB exempt as the cells were commercially purchased. Cells were passaged when they reached 85% confluency using TrypLE Express (Life Technologies, Carlsbad, CA, USA) and plated into six-well or 10-cm dishes (Corning, St. Louis, MO, USA) at 4,500 cells/cm2 for SNP genotyping or Western blot analy sis, respectively. TALE-KRAB or TALE-FokI DD and TALE-FokI RR fusion vectors were cotransfected into HD patient-derived fibroblast cells (GM02151) with an eGFP reporter plasmid under a CMV promotor (Addgene). Transfections were performed via two different kits: Lipofectamine 3000 and polyethylenimine (PEI) following the manufacturer's protocol.
Viability Assay
To measure toxicity and cell death following administration of the TALE, we used well-validated flow cytometry methods to quantify the percentage of viable cells. Cells were passaged 48 h after transfection and costained with annexin V and propidium iodide (PI) per the Annexin V-FITC Apoptosis Kit (Invitrogen, Carlsbad, CA, USA). Cells were counterstained with Hoescht 33348 (1:1,000; Thermo Fisher Scientific, Waltham, MA, USA) for 15 min at room temperature. Viability was determined via FACS (FC500; Beckman-Coulter, Brea, CA, USA). Twenty thousand events were recorded for each condition.
Western Blot
To semiquantify the aggregates associated with the mutant huntingtin protein, we performed Western blot experiments using a previously validated antibody for human HD fibroblasts (14). Protein was extracted 2 days posttransfection via standard protein isolation protocol using Pierce RIPA buffer (Thermo Fisher Scientific) with a halt protease & phosphatase inhibitor cocktail (Thermo Fisher Scientific). Protein lysate (10 mg) was loaded into wells of an SDS-polyacrylamide gel (Life Technologies) and were electrophoresed (Life Technologies) for 1 h at 200 volts. Protein was transferred onto prepared PVDF membrane (Life Technologies) by first equilibrating the gel in transfer buffer for 15 min and then transferring the gel onto the PVDF membrane running at 30 volts overnight at 4°C. Membranes were blocked with 5% nonfat milk (Bio-Rad, Hercules, CA, USA) for 1 h at room temperature. The membrane was then transferred to a solution containing anti-ubiquitin (1:500, MAB1510; Millipore, Temecula, CA, USA) and 5% BSA (Bio-Rad) in TBS (Bio-Rad) overnight at 4°C with gentle agitation. Membrane was washed with TBST (Thermo Fisher Scientific, Pittsburg, PA, USA) three times before incubation with anti-mouse IgG-HRP (1:1,000, SC-2005; Santa Cruz Biotechnologies, Santa Cruz, CA, USA) for 1 h at room temperature. Bands were developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) for 5 min at room temperature. Membranes were imaged on a ChemDoc XRS+ (Bio-Rad) using ImageLab 5.1 software (Bio-Rad). The same procedure was then performed using anti-β-actin (1:1,000, A5441; Sigma-Aldrich, St. Louis, MO, USA) as a housekeeping protein after stripping the membrane for 15 min at room temperature using Restore PLUS Western Blot Stripping Buffer (Thermo Fisher Scientific).
SNP Genotyping Assay
The SNP genotyping assay from Life Technologies enables the quantification of transcription levels of individual alleles using a distinct primer probe set synthesized to identify a SNP between the two alleles. RNA extractions were performed 2 days posttransfection using RNEasy Plus mini kit (Qiagen, Hilden, Germany). cDNA synthesis was subsequently performed using the iScript cDNA Synthesis kit (Bio-Rad). SNP genotyping assay was performed using TaqMan Probes for SNP rs363070 as it exists in the 3’ end of the mutant huntingtin RNA and has been previously validated by Life Technologies. SNP genotyping following our protocol allows for the quantification of the transcription of both the mutant and healthy huntingtin alleles using Q-PCR. The manufacturer's protocol was followed with the exception of using cDNA. Input cDNA (10 ng) from each treatment group underwent PCR in triplicate. Genotyping assays were performed in the StepOnePlus PCR system (Applied Biosystems, Foster City, CA, USA) and analyzed in StepOne v2.2.2 (Applied Biosystems).
GFP Cell Sorting
To measure the potency of the TALE in only fibroblasts that were successfully transfected, we performed a fluorescent activated cell sorting experiment to purify the cells based on a fluorescent tag incorporated into the TALE plasmid. HD fibroblasts were transfected with TALE-fused KRAB with eGFP using the same protocol described above. Forty-eight hours posttransfection, cells were sorted by fluorescent activated cell sorting (FACS; UC Davis FACS Core). Cells were sorted in PBS using BD Influx Cell Sorter (BD Biosciences, San Jose, CA, USA) a five-laser system consisting of a blue laser (100 mW at 488 nm), red laser (30 mW at 635 nm), green laser (150 mW at 532 nm), violet laser (50 mW at 405 nm), and UV laser (100 mW at 355 nm). The instrument was set up with the 100-μM nozzle and aligned on day of sort. eGFP-positive and -negative cells were sorted and immediately plated as described above. Twenty-four hours after FACS, the cells underwent SNP genotyping as described above.
Statistical Analysis
All statistical analysis was performed using SPSS v22. Multivariate or one-way analysis of variance (ANOVA) was used to compare between-group differences with least significantly different (LSD) post hoc analysis performed when appropriate. All data are presented as mean and standard error.
Results
All TALEs were sequence verified for the correct promoter, repeat variable diresidue (RVD), and effector domain (Fig. 1). The ability of the TALE to bind to its target sequence was assessed using a Luciferase binding assay (3). TALEs T7, T2, T3, CAG-F, and CAG-R were cloned with a transcriptional activator (VP64) and cotransfected into HEK293T cells with a reporter plasmid containing the respective TALE binding sites upstream of a luciferase gene. All TALEs constructed demonstrated luciferase expression when compared to control transfections (Fig. 2A). Data are presented as luciferase expression over the respective target sequence without TALE and controlled for transfection efficiency. All TALEs demonstrated equivalent expression as the positive control cells transfected with the luciferase gene under the normal pGL3 promoter.

Transcription activator-like effector (TALE) design. The repeat variable diresidue (RVD) sequence can be seen by the colored lettering. The target sequence is in black text. For single-nucleotide polymorphism (SNP) targeting, the allele mismatch can be seen in the outlined box. The effector domain for each plasmid is shown in the right column.
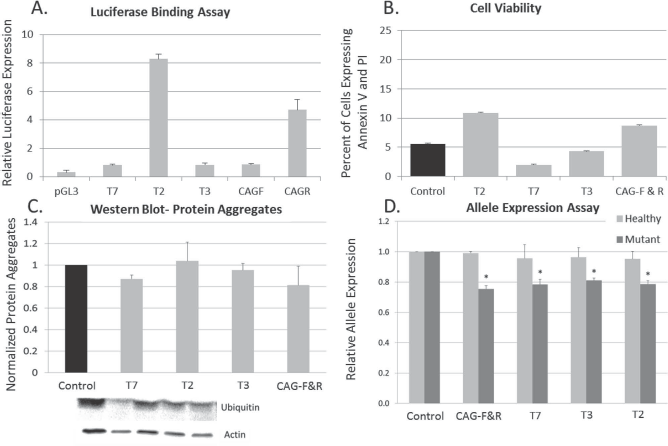
TALE binding assay. (A) TALE binding was assessed using a luciferase-binding assay in which the TALE was cloned with an activator effector domain (VP64) and cotransfected into HEK293 cells with a luciferase reporter plasmid containing the TALE target region. All TALE constructs demonstrated luciferase expression when compared to control transfections (n = 4). (B) Toxicity was measured 48 h posttransfection with Lipofectamine 3000 by flow cytometry with annexin V fluorescein isothiocyanate (FITC) and propidium iodide (PI). Cells were considered viable if they were negative for annexin V/PI. No differences were observed in toxicity between the phosphoglycerate kinase (PGK) empty vector and the TALE targeting SNP sites or CAG regions (n = 3). (C) Protein aggregation was quantified with a Western blot for anti-ubiquitin. No overall between-group differences were observed in the expression of ubiquitin protein aggregation [F(8, 26) = 0.512, p = 0.832] (n = 5). (D) Allele expression was quantified using a Life Technologies SNP genotyping assay. One-way ANOVA revealed no significant between-group differences in expression of the healthy allele [F(4, 18) = 0.994, p = 0.443], suggesting that the TALE does not target the healthy allele. A significant between-group difference was observed for expression of the mutant allele [F(4, 18) = 3.021, p = 0.05]. LSD post hoc analysis revealed a significant difference in mutant allele expression between control (pPGK empty) and CAG-FR (p = 0.007), T7 (p = 0.009), T3γ (p = 0.021), and T2 (p = 0.010). LSD post hoc analysis revealed a significant difference between mutant and healthy allele expression in CAG-FR (p = 0.001), T3γ (p = 0.05), T2 (p = 0.022), and a trend in T7 (p = 0.061) (n = 6). *Significant from Control.
The potential toxicity of the TALEs was tested using a cell viability assay. Briefly, toxicity was measured 48 h posttransfection of TALEs into HD fibroblasts by flow cytometry with annexin V and PI. Cells were considered viable if they did not have annexin V or PI signal. No differences were observed in toxicity between the pPGK-empty vector and the TALEs targeting SNP sites or CAG regions (Fig. 2B).
Protein aggregation was quantified with a Western blot for anti-ubiquitin. The ubiquitin-proteasome system has been implicated in HD progression and is used as a marker of disease progression (44). No overall between-group differences were observed in the expression of ubiquitin protein aggregation [F(4, 14) = 1.343, p = 0.320]; however, a trend toward reduced protein aggregation was observed in several of the TALEs (Fig. 2C).
Allele expression was quantified using a SNP genotyping assay. One-way ANOVA revealed no significant between-group differences in expression of the healthy allele [F(4, 17) = 1.134, p = 0.383], suggesting that the TALE does not target the healthy allele. A significant between-group difference was observed for expression of the mutant allele [F(4, 17) = 3.889, p = 0.027]. LSD post hoc analysis revealed a significant difference in mutant allele expression between control (pPGK empty) and CAG-F&R (p = 0.007), T7 (p = 0.009), T3 (p = 0.021), and T2 (p = 0.010). These data suggest that TALEs designed to cause either CAG collapse or gene silencing via SNP targeting are potent in selectively reducing mutant allele expression (Fig. 2D).
Transfection efficiency was measured using immunocytochemistry and revealed that 15–23% of the cells were transfected and expressed eGFP (Fig. 3A). Correlational analysis from the same experiment revealed that the number of cells transfected had high negative correlation with the expression of the mutant allele and protein aggregation, suggesting that the TALE is highly potent when delivered to the cells (data not shown). Fluorescent activated cell sorting (FACS) to purify only the cells receiving TALEs demonstrated approximately 68% reduction of the mutated allele 24 h after sorting (Fig. 3B), suggesting the TALE is highly potent, and the delivery of the therapeutic needs to be optimized. FACS purification of the cells demonstrated the potency of the TALEs; however, due to the low transfection efficiencies and limited samples FACS could not be performed for all analyses.

Transfection efficiency was measured using fluorescent microscopy. (A) All TALE transfections resulted in approximately 15–23% efficiency. (B) Fluorescent activated cell sorting to purify only the cells receiving TALE demonstrated approximately 68% reduction of the mutate allele 24 h after sorting (n = 3). GFP, green fluorescent protein.
Discussion
The results from this study indicate that transcription activator-like effectors can be constructed and utilized to reduce mutant allele expression in human HD-derived fibroblasts. Multiple TALEs were created to target SNP sites occurring in or near the promoter region of the huntingtin gene and paired with the transcriptional repressor KRAB. A second set of TALEs were created to induce a collapse of the CAG repeat by using heterodimeric nuclease FokI in a paired fashion to induce a double-stranded break of the DNA only in CAG lengths containing greater than 15 repeats. All of the TALEs showed efficient binding in the luciferase assay as demonstrated by expression of the luciferase gene above the levels observed in the control cells. These results verify the construction of the TALE and, more importantly, the ability of each construct to bind and regulate gene expression. Each TALE or combination of TALEs did not demonstrate cell toxicity above what was observed in the control transfected cells, confirming that the TALE itself is not inherently toxic to cells.
Western blot analysis revealed a trend toward decreased protein aggregation in the T7-, T3-, and GAG-F&R-treated Huntington fibroblasts compared to cells treated with empty vector. These trends correlated well with the transfection efficiency (described below). Western blot is a less sensitive measure than the SNP genotyping assay but still provides valuable insight into the reduction of mutant protein. It is likely that the large number of non-transfected cells skews the results and limits the statistical power of this measure.
The transcriptional repressor TALE T7, T3, and T2 demonstrated significant mutant allele knockdown as measured by the SNP genotyping assay. Furthermore, none of the TALEs tested in this study reduced expression of the healthy allele, demonstrating allele specificity of the gene regulation approach. Significant reduction of mutant allele expression of upward to 20% was observed with TALE paired with KRAB that was designed to target to SNPs occurring in the mutant allele in a mixed population of treated and untreated fibroblasts. Interestingly, a significant reduction of mutant allele expression was observed in HD fibroblasts treated with CAG-F and CAG-R, which were designed to create a double-stranded break, collapse of the CAG region, and allele repair without interfering with allele expression. It is possible that the hypothesized DNA repair mechanism in HD is impaired and the normally efficien single-strand annealing repair pathway was disrupted (16), resulting in lowered allele expression. The level of mutant allele repression that will be required to significantly alter or halt disease progression is not yet known. Further experiments were performed to determine whether the TALE was approximately 20% efficient at reducing expression of the mutant huntingtin allele or whether traditional transfection techniques limited the effectiveness of the TALE. It has been previously reported by other groups that fibroblasts are difficult to transfect cells and have low transfection efficiencies (18), similar to our findings.
The potency of the TALEs was further validated through correlational and cell-sorting experiments. In the first set of experiments, the TALE plasmids were cotransfected with a plasmid containing eGFP under a CMV promoter. The cells were then imaged to quantify the percentage of HD fibroblasts that had received the TALE. It was found that transfection efficiency of TALE and eGFP plasmids into fibroblasts was 15–23%, similar to previous reports (18). However, many of the TALEs displayed a strong negative correlation between transfection efficiency and mutant allele reduction and transfection efficiency and protein aggregation, demonstrating that the transfection may be the limiting factor in mutant allele reduction (data not shown).
In an attempt to characterize the potency of our TALE allele regulation platform, T7, a TALE targeted to a SNP and paired with a transcriptional repressor that showed significant reduction and allele specificity, was recloned and paired with eGFP following KRAB. This plasmid was then transfected into HD fibroblasts and the eGFP-positive and eGFP-negative cells were sorted via fluorescence activated cell sorting, replated for 24 h to allow the cells to recover without undergoing more than one population doubling. These cells were then analyzed with the SNP genotyping assay. Transfection efficiency was similar to that observed in the correlational study. Isolation of an eGFP-positive population revealed reduced expression of the mutant huntingtin allele by approximately 68%. While this experiment did not demonstrate complete silencing of the mutant allele, it is possible that a small percentage of non-GFP cells were cultured or that a small percentage of the cells proliferated and that the daughter cells did not contain the TALE. More importantly, these data suggest that TALEs are highly potent and efficient in reducing the expression of the mutant huntingtin allele.
Although the different TALEs constructed did not display the same efficacy, these results will help future studies design their gene-editing approaches around target sites that displayed better efficiency (i.e., the location of the SNP within the promoter and where the SNP falls within a region of high specificity for the TALE). The relatively quick design and validation of multiple TALEs allows the flexibility for allelic silencing, pending SNP allele analysis, or CAG collapse, as potential gene therapies, thus potentially providing therapy to a large proportion of the HD and JHD populations (Fig. 4).
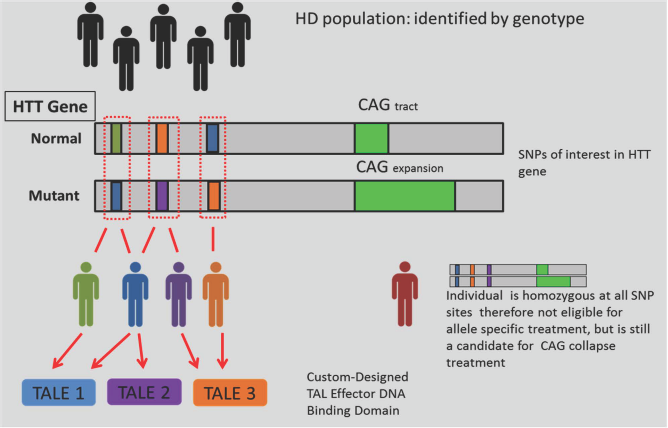
Application to the patient population. In theory, allele-specific targeting using TALE could potentially cover 100% of the patient population. The use of multiple TALEs may be capable of covering 100% of the patient population, allowing flexibility for either allelic silencing, pending SNP allele analysis, or CAG collapse in regards to potential gene therapies. HTT, Huntingtin gene. Adapted from Kay et al. (22).
While these results demonstrate great promise in terms of targeted allele manipulation, much work will be needed to demonstrate the preclinical efficacy and safety of this new approach. Future studies should focus on characterizing an appropriate delivery system for in vivo administration of the most effective TALE into transgenic mouse models of HD. Similar studies to those described here could also be performed in HD and JHD patient-derived neurons to demonstrate that the TALE is efficient in reducing allele expression without overt toxicity, similar to what was observed in primary patient fibroblasts. Characterization of patient-derived neurons could also be performed to validate the ability of the TALE to restore normal synaptic function as well as other downstream disease phenotypes, such as mitochondrial damage and the accumulation of reactive oxygen species.
Footnotes
Acknowledgment
This work was supported by a National Institute of Health National Research Service Award Postdoctoral Fellowship (F32NS090722) (K. D. Fink), Graduate Fellowships (NSF GRFP 011116000, NIH T32-GM008799, NSF GROW 201111600, T32-HL086350) (J. D. Anderson), NIH NIGMS Predoctoral Fellowship (T32GM099608) (P. Deng), National Institute of Health Director's transformative award (R01GM099688) (J. A. Nolta), National Institute of Health (R01GM097073) (D. J. Segal), California Institute for Regenerative Medicine (CIRM DR2-05415) (V. Wheelock/J. A. Nolta), Stewart's and Dake Family Gift (K. D. Fink), Help4HD International, the University of California Davis Flow Cytometry Shared Resource Laboratory with funding from the NCI P30 (CA0933730), NIH (NCRR C06-RR12088, S10 RR12964 and S10 RR 026825) with technical assistance from Ms. Bridget McLaughlin and Mr. Jonathan Van Dyke, and philanthropic donors from the HD community, including the Roberson family and TeamKJ. The authors declare no conflicts of interest.