
Abstract
Establishing an efficient differentiation procedure is prerequisite for the cell transplantation of pluripotent stem cells. Activating fibroblast growth factor (FGF) signals and inhibiting the activin/nodal pathway are both conserved principles to direct the neural induction (NI) of developing embryos and human embryonic stem cells (hESCs). Wnt signal and OCT4 expression are critical for the hESC pluripotency; however, their roles in cell differentiation are largely unclear. We demonstrate that in the presence of FGF2 and activin inhibitor SB431542, applying a small-molecule Wnt agonist, BIO, efficiently and rapidly steers the NI of all our tested hESCs. A human induced pluripotent stem cell (iPSC), which is refractory for efficient neural conversion by FGF2, effectively differentiated to SOX1+ cells after the BIO/SB431542/FGF2 treatment. In addition, BIO promoted cell survival and transiently sustained OCT4 expression at the early NI stage with FGF2 and SB431542. Interestingly, at the late NI stage, the OCT4 level rapidly declined in the treated hESCs and consequently initiated the formation of neural rosettes with forebrain neuron characteristics. This study illustrates the distinct effects of Wnt activation on maintaining pluripotency and committing neural lineages at the early and late NI stages of hESCs and iPSCs, respectively.
Introduction
Neural induction (NI), the stage in which pluripotent stem cells are converted into neural progenitor cells (NPCs), has been extensively investigated in developing vertebrate embryos (12,38). Blockage of transforming growth factor-β (TGF-β) superfamily activation, such as bone morphogenetic proteins (BMPs) and activin/nodal signals, is necessary for the initiation of NI in the embryo, and this principle is also recapitulated during the neural formation of mouse and human embryonic stem cells (mESCs and hESCs) (11,29). For example, providing both noggin protein and the SB431542 small molecule, a BMP antagonist and activin receptor inhibitor, respectively, could efficiently and rapidly trigger the NI of hESCs (5), which benefits the further application of hESCs on basic neurosciences and cell transplantation.
In addition to TGF-β inhibitors, activated fibroblast growth factor (FGF) signals potentiate the NI in vertebrates and ESCs (6,42). Treating H9 hESCs with FGF2 alone in the absence of fetal bovine serum effectively converts hESCs into NPCs (42). Mechanistic investigations of FGF-triggered NI illustrate that FGF2 provides a BMP-independent neurogenic signal for steering the neural rosettes formation of differentiating H9 hESCs (20).
Notably, the high NI efficiency using FGF2 is restricted to limited hESC lines, but not reproduced in some hESCs and induced pluripotent stem cells (iPSCs) (16). These observations suggest that unidentified differentiating signals may be required for the efficient NI of pluripotent stem cells. Accumulating evidence indicates that substantial differences in imprinting genes and epigenetic configuration exist among different hESCs (1,4). In addition, iPSCs reprogrammed by introducing Yamanaka factors still partially maintain the methylated epigenetic tags of original somatic cells in their genomes (18,19). These results suggest that the status of pluripotent gene expression among differentiating hESCs and iPSCs may be substantially varied and may lead to different efficiencies of neural conversion for hESCs and iPSCs.
Whether wingless-type mouse mammary tumor virus (MMTV) integration site (Wnt) signaling is required for maintenance of the self-renewal of pluripotent stem cells or promoting the differentiation remains controversial (9,28). The 6-bromoindirubin-3′-oxime (BIO) small molecule, a well-characterized Wnt agonist (27), has been demonstrated to sustain the pluripotent status of hESCs (35). We applied BIO with two neural differentiating factors, FGF2 and SB431542, from differentiation day 3 (D3) to D4. Surprisingly, transient BIO treatment did not block hESC differentiation, but significantly potentiated neural rosette formation in all of the tested hESCs and iPSCs. The molecular profiles of the ESC-derived neural cells were examined, including markers of NPCs, regional specification, and the competence of neural patterning. The underlying mechanisms of BIO-mediated NI enhancement and its relationship with octamer-binding transcription factor 4 (OCT4) expression were further investigated in this study.
Materials and Methods
hESC and iPSC Cultures
The hESC lines, including H1 (XY, passages 75–85; WiCell, Madison, WI, USA), H9 (XX, passages 45–55; WiCell), TW1 (XX, passages 80–90), and hESC6 (XY, passages 70–80) were grown in mTeSR1 media (Stem Cell Technologies, Vancouver, BC, Canada) on 1.0% Matrigel (Becton Dickinson, BD, Franklin Lakes, NJ, USA)-coated 6-cm dishes (Corning, Corning, NY, USA). The TW1 cells (8) and hESC6 (or named as T5) cells (21) were both reported and transferred from Lee Women's Hospital and Profertile IVF Center in Taichung, Taiwan, respectively. Two iPSCs, including IMR90-1 (XX, passages 67–75; WiCell) and amniotic fluid-derived iPSCs (AF-iPSCs, XX, passages 23–30; Food Industry Research and Development Institute, Hsinchu, Taiwan) (24), were grown under the same culture condition. The culture medium was refreshed daily. The cells were passaged using dispase II (0.5 mg/ml; Invitrogen, Carlsbad, CA, USA) and replated at a cell dilution of 1:5 to 1:8.
Neural Induction
After being treated with dispase II for 5 min, the detached hESCs and iPSCs (1.0 × 106 cells) were partially dissociated into 200–300-μm cell clusters and transferred to 6-cm bacterial culture dishes (Alpha Plus Scientific Corp., Taoyuan, Taiwan) for 2-day culture. The differentiating medium contained Dulbecco's modified Eagle's medium/F12 (DMEM-F12; Invitrogen) and 20% knockout serum replacement (KSR; Invitrogen), 1 mM nonessential amino acids (NEAAs; Invitrogen), 2 mM glutamate (Invitrogen), and 0.1 mM 2-mercaptoethanol (Invitrogen). The day of cell dissociation was set as D1. From D3 to D4, the cell suspensions were cultured in DMEM-F12 with 1% N2 supplement (Invitrogen), 1 mM NEAAs, and 2 mM glutamate. Neural-inducing factors were applied at this D3–D4 stage, including 10 μM SB431542 (Sigma-Aldrich, St. Louis, MO, USA), 0.5 μM BIO (Sigma-Aldrich), and/or 10 ng/ml recombinant human FGF2 (rh-FGF2; R&D Systems, Minneapolis, MN, USA). From D5, the neural-inducing factors were removed, and the cells were further cultured in neurobasal media (Invitrogen) with 1% N2 supplement and 10 ng/ml rh-FGF2.
Motor Neuron and Dopaminergic Neuron Induction
The procedure for motor neuron and dopaminergic neuron induction was modified from previous reports (22, 25,34). Briefly, the NPCs were adhered to 1% Matrigelcoated plates on D10. Motor neurons were induced by the treatment with 0.5 μM retinoic acid (RA; Sigma-Aldrich) and 1 μM purmorphamine (Merck-Millipore, Billerica, MA, USA), a sonic hedgehog (Shh) signal activator (23), from D11 to D15. The formation of dopaminergic neurons was induced by the addition of 40 μg/ml FGF8b (R&D Systems) and 1 μM purmorphamine for 5 days. The medium was refreshed every 2 days. To promote terminal neural differentiation, a cocktail containing 10 ng/ml brain-derived neurotrophic factor (BDNF; Peprotech, Rocky Hill, NJ, USA), 10 ng/ml glia cell line-derived neurotrophic factor (Peprotech), and 5% B27 supplement (Invitrogen) in neurobasal media was provided from D16. Motor neurons and dopaminergic neurons were examined by immunocytostaining on D25.
Flow Cytometry Analysis
Cells were washed twice with phosphate-buffered saline (PBS; Sigma-Aldrich) and were permeabilized with BD permeabilizing solution 2 (BD) for 2 min. Cellular antigens were blocked with 5% horse serum before the staining of primary antibody. We incubated OCT4 (1:500; Santa Cruz Biotechnology, Dallas, TA, USA) and nestin (1:500; Convance, Princeton, NJ, USA) antibody at 4°C for 30 min and used Alexa Fluor 488 (Invitrogen)-labeled secondary antibodies for quantification. Cells were analyzed by BD Influx cell sorter, and the ratio of fluorescent-positive cells was estimated with FlowJo software (Ashland, OR, USA).
Immunocytochemistry (ICC) Staining
Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 5 min and washed twice with PBS (Sigma-Aldrich). After membrane permeabilization with 0.3% Triton X-100 (USB, Cleveland, OH, USA), cellular antigens were first blocked with 5% horse serum (Invitrogen) and then stained with primary antibodies at 4°C overnight. The primary antibodies used in this study included OCT4 (1:500; Santa Cruz Biotechnology), sex-determining region Y box 2 (Sox2, 1:500; Millipore), Nanog (1:500; Reprocell, Yokohama, Japan), Brachyury (T, 1:50; Santa Cruz Biotechnology), Sox17 (1:20; R&D Systems), Sox1 (1:200; Santa Cruz Biotechnology), paired box 6 (Pax6, 1:100; Covance), zinc finger of the cerebellum 1 (Zic1, 1:200; Abcam, Cambridge, MA, USA), nestin (1:500; Covance), N-cadherin (1:500; Santa Cruz Biotechnology), brain factor 1 (BF-1, 1:200; Development Studies Hybridoma Bank, DSHB, Iowa City, IA, USA), neuronal precursor cells (Forse-1, 1:50; DSHB), zona occludens 1 (ZO-1, 1:100; Zymed-Invitrogen), homeobox B4 (HoxB4, 1:50; DSHB), oligodendrocyte lineage transcription factor 2 (Olig2, 1:50; Santa Cruz Biotechnology), motor neuron and pancreas homeobox 1/homeobox HB9 (MNX1/HB9, 1:50; DSHB), tyrosine hydroxylase (TH, 1:100; Covance), βIII-tubulin (clone TuJ1, 1:500; Covance), and glial fibrillary acidic protein (GFAP, 1:500; Millipore). Cell nuclei were stained with diamidino-2-phenylindole (DAPI; Invitrogen). Fluorescent cell images were obtained using an upright microscope (Eclipse TE2000-S and 80i; Nikon, Tokyo, Japan) or a confocal microscope (LSM 510; Carl Zeiss, Oberkochen, Germany). The population ratios of specific protein-expressing cells were estimated by manual counting with AlphaEase FC software (Alpha Innotech, San Leandro, CA, USA).
Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Total RNA from differentiating ESCs was extracted and then converted to cDNA using reverse transcriptase (Invitrogen). The cDNA was used as a template for realtime PCR using ProTaq™ DNA polymerase (Protech Technology Enterprise Co., Ltd., Taipei, Taiwan) supplemented with SYBR Green I (Cambrex, East Rutherford, NJ, USA). The conditions for the PCR reaction included the following steps. The DNA templates were denatured for 5 min at 94°C, followed by 40 repeated cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The forward OCT4 primer is 5′-GGT ATT CAG CCA AAC GAC CA-3′, and the reverse primer is 5′-CAC GAG GGT TTC TGC TTT GC-3′. The expression of histone H2, a housekeeping gene, was as an internal control of RT-PCR. The forward histone H2 primer is 5′-TAG CCT TTT CTC TGC CTT GC-3′, and the reverse primer is 5′-TGG ATG TGT GGA ATG ACA CC-3′. The PCR sizes of OCT4 and histone H2 genes are 151 bps and 370 bps, respectively. The real-time quantitative PCR was performed using a Rotor-Gene 3000 (Qiagen-Corbett Hilden, Germany), and the obtained data were analyzed with the installed Rotor-Gene6 software.
Production and Infection of Recombinant Lentiviruses
The recombinant green fluorescent protein (GFP) and OCT4-shRNA lentiviruses were generated by transient transfection with the pMD2.G, psPAX.2 packaging plasmid, and the shOCT4 pLKO.1-puro plasmids in human embryonic kidney 293 T (HEK293T) cells (gifted from Professor Ming-Hong Tai at National Sun Yat-sen University, Taiwan). All of these plasmids were obtained from National RNAi Core Facility Platform at Academia Sinica, Taiwan. The harvested HEK293T cell media were centrifuged at 300 × g for 5 min at room temperature, and the supernatants were purified through a 0.45-μm (Merck-Millipore) filter to remove the cell debris. The filtered media were transferred to polyallomer centrifuge tubes (Beckman-Coulter, Brea, CA, USA) and ultracentrifuged at 24,000 rpm in a SW28 rotor (radius 161 mm; Beckman-Coulter) for 90 min at 4°C. The harvested viral pellets were resuspended in DMEM and titrated by puromycin (Sigma-Aldrich) selection in HEK293T cells. The differentiating hESCs were infected with the recombinant lentiviruses at a multiplicity of infection (MOI) of 5 for 24 h at 37°C in the presence of 5 μg/ml polybrene (Sigma-Aldrich).
Statistical Analysis
Representative data were collected from at least three independent experimental results and shown as mean value ± standard deviation. Statistical analyses were conducted using one-way ANOVA (Excel; Microsoft, Redmond, WA, USA), and the significance was examined by Tukey's post hoc assay (Prism 5; Graph Pad, San Diego, CA, USA).
Results
Robust Neural Formation of TW1 hESCs by Treating with BIO/SB431542/FGF2
The differentiation schedule of this study is illustrated in Figure 1A. Undifferentiated TW1 hESCs (Fig. 1B I) were cultured in nonserum, nonfeeder, and chemically defined mTeSR1 medium to expand the cells reproducibly and to avoid potential interference of feeder cells on NI. After cell dissociation with dispase, suspended TW1 hESCs were first adapted in mTeSR1 medium for 12 h. The culture medium was then replaced with DMEM-F12 containing 20% KSR for forming embryoid bodies and steering cell differentiation. The cells were transiently treated with 0.5 μM BIO/10 μM SB431542/10 ng/ml FGF2 (BiSF) on D3 for 2 days and further cultured with 10 ng/ml FGF2 in neurobasal medium with N2 supplement. Compact and donut-like cell aggregates were found on D6 (Fig. 1B II). A significant number of the differentiating hESCs consistently displayed a classical neural rosette conformation after plating on culture plates on D8 (Fig. 1B III). The neural cell lineages were further confirmed by the expression of specific neural markers, including SOX1, PAX6, and ZIC1 transcription factors on D10 (Fig. 1C I–III). The acquired cell polarity of the NPCs was illustrated by the lumen formation of columnar rosettes and the intense expressions of N-cadherin and ZO-1 on the circular margins of the lumens on D15 (Fig. 1D I, II). Individual neural rosettes, featured with membranous N-cadherin and nuclear ZIC1 expression, exhibited the diameter of the rosettes around 100 μm (Fig. 1D III).

BiSF treatment steered efficient NI of TW1 hESCs. (A) The schematic procedure for the BiSF condition is illustrated. (B) Phase contrast images illustrated the undifferentiated human embryonic stem cells (hESCs), suspended embryoid bodies on day 6 (D6), and adherent columnar early neural rosettes on D8. (C) BiSF-induced neural progenitor cells (NPCs) expressed the neural markers, such as sex-determining region Y box 1 (SOX1), paired box 6 (PAX6), and zinc finger of the cerebellum 1 (ZIC1) on D10. (D) The inner lumens of the neural rosettes were delineated by the N-cadherin and zona occludens 1 (ZO-1) expressions. Individual neural rosettes were visualized by the N-cadherin and ZIC1 staining. KSR, knockout serum replacement; DMEM, Dulbecco's modified Eagle's medium; BiSF, 0.5 μM 6-bromoindirubin-3′-oxime (BIO)/10 μM SB431542/10 ng/ml fibroblast growth factor 2 (FGF2); NI, neural induction; N-cad, N-cadherin. DAPI (4′,6-diamidino-2-phenylindole) was used as a nuclear stain. Scale bar: 100 μm (B I–II and D III), 50 μm (C I–III and D I, II).
The kinetics and efficiency of NI by BiSF treatment were revealed by the ICC staining. The pluripotent OCT4 proteins were expressed in 33.5 ± 3.7% of the differentiating TW1 hESCs on D5 (Fig. 2A I), but only 9.5 ± 2.3% of the cells were detected on D8 (Fig. 2A II) and barely detected on D10 (Fig. 2A III). Nestin, a well-characterized NPC marker, was first detected on D5 and robustly expressed in BiSF-steered hESCs on D8 and D10 (Fig. 2A). On D10, the BiSF-treated cells expressed other specific NPC markers, such as SOX1 (82.5 ± 3.0%), PAX6 (68.5 ± 2.4%), and ZIC1 (74.5 ± 0.5%) (Fig. 2B, C). One interesting finding is that ZIC1, a general neural transcription factor (3,26), was detected on D5, as early as the emergence of nestin (Fig. 2C). This evidence strongly argues that the BiSF protocol is a rapid and highly efficient NI method.
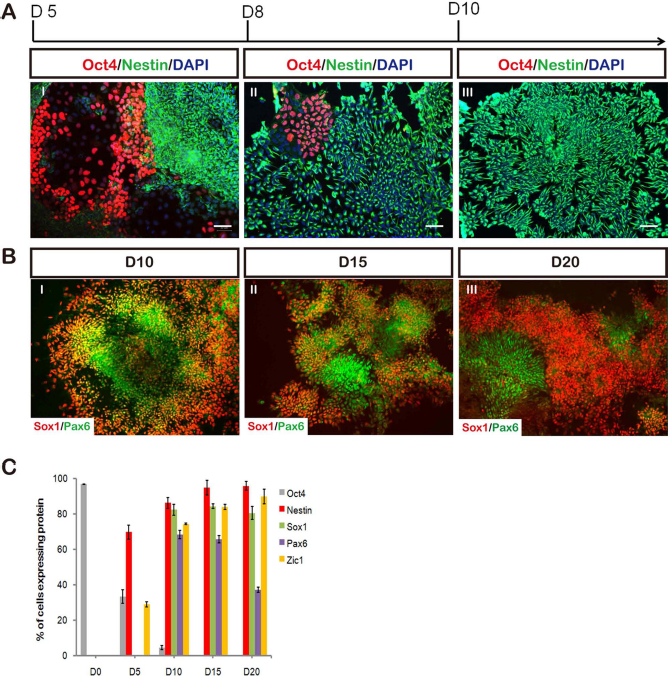
The kinetic expression of neural markers under the BiSF condition. (A) The pluripotency of hESCs was evaluated by the ocatmer-binding transcription factor 4 (OCT4) expression, while the neuralization was marked by nestin on D5, D8, and D10. (B) The SOX1 and PAX6 transcription factors were quantified by the immunocytochemistry (ICC) staining on D10, D15, and D20. (C) The ratios of the protein expressions from D5 to D20 were summarized and plotted. Each data represented three independent results. Scale bars: 50 μm.
During the neural development of mouse embryos, the SOX1 transcription factor is expressed prior to PAX6 in the primitive neural epithelium (31). This prior expression of SOX1 is recapitulated in the neural differentiation model of mESCs (39), but not in that of hESCs (43). Notably, on D8, 82.2 ± 2.1% of the differentiating cells were marked with SOX1; however, only a few cells were labeled with PAX6. These PAX6-expressing cells reached 68.5 ± 2.4% of the total population on D10 and gradually declined to 37.3 ± 1.4% of the cells on D20 (Fig. 2B, C). The PAX6 expression pattern almost completely colocalized with the SOX1+ cells on D10 and D15 (Fig. 2B I, II) but was detected in separate SOX1-null neural populations on D20 (Fig. 2B III). We also noticed that compared to the D3 to D4 (2 days) treatment (Fig. 3A), treating the TW1 hESCs with BiSF from D3 to D6 (4 days treatment, Fig. 3B) significantly abolished the expression of PAX6 (68.5 ± 2.4% to 21.3 ± 6.4%, p < 0.01), but not the other neural markers, such as SOX1, nestin, and N-cadherin. This result suggests that during the NI stage, Wnt activation may trigger the SOX1 expression, but conversely prohibit the PAX6 expression. These results also indicate that the molecular patterns of NI in hESCs under the BiSF culture condition, especially for the order of SOX1 and PAX6 expressions, are similar to those of neural formation in mouse embryos and mESCs.

The effects of duration of BiSF treatments on SOX1 and PAX6 expression. (A) The TW1 hESCs were treated with BiSF on D3–D4 (2 days) or (B) treated on D3–D6 (4 days). The cells were fixed and stained with indicated antibodies on D10. Scale bar: 50 μm.
Comparison of the NI Efficacy Under FGF2, SB431542/FGF2, and BiSF Conditions
To compare the NI efficacy with FGF2, SB431542/ FGF2 (SF), or BiSF, the cell fates of differentiating hESCs were investigated with NPC markers, such as the N-cadherin, ZIC1, SOX1, and PAX6. A mature neuron marker, βIII-tubulin (antibody clone TuJ1), was also included. In addition, classical markers of the mesoderm and endoderm, such as Brachyury (T) and SOX17, respectively, were also evaluated. The results in Figure 4A reveal that the FGF2- or SF-treated cells showed moderate cell death and few neural rosettes during the NI (Fig. 4A I, II), whereas the BiSF treatment steered the majority of the TW1 hESCs to form extensive neural rosettes on D15 (Fig. 4A III). These results indicate that the NI of TW1 cells was less efficient when only FGF2 or SF is provided within D3 to D4. For example, for the FGF2-treated TW1 cells, approximately half of the cell population was still restricted at the pluripotent stage (OCT4+ cells) on D15 (Fig. 4B I), and very few cells were labeled with SOX1 or ZIC1 (Fig. 4C I, D I). Only 5.2 ± 3.6% of the PAX6+ cells and 10.1 ± 5.1% of the N-cadherin+ cells were detected in the FGF2-treated cells (Fig. 4E). Among the SF-treated hESCs, 36.3 ± 4.4% and 42.7 ± 1.3% of cells expressed the SOX1 and ZIC1 transcription factors, respectively (Fig. 4C II, D II, E). In contrast, the BiSF treatment robustly enhanced the neural cell conversion, evidenced by the extensive expressions of SOX1, ZIC1, and N-cadherin (80 ± 4.7%) (Fig. 4C III, D III, E). These BiSF-treated hESC progeny also exhibited extensive neurite formation and terminal differentiation on D15 (clone TuJ1+, 63.2 ± 6.0%). Exploring the nonneural lineages by ICC showed that less than 10% of cells differentiated into endomesodermal cells (T+ and SOX17+ cells) under these three NI conditions (Fig. 4E). The low yield of endomesodermal cells may be caused by the nonpermissive environment for the differentiation or growth of endomesodermal cell lineages in the neurobasal medium.

The NI efficacy of TW1 hESCs under the FGF2, SF, or BiSF condition. (A) Representative phase contrast images of the differentiating hESCs on D15 are provided. (B–D) The expressions of OCT4 (B), SOX1 (C), and ZIC1 (D) were detected in the hESC progeny on D15 using the individual NI methods. (E) The statistical analyses of the protein expressions were summarized from triplicate ICC staining results. Tuj1, antibody clone for βIII-tubulin; T, Brachyury. Scale bar: 100 μm (A I–III), 50 μm (B–D).
Neural Formation of other hESCs and iPSCs
To evaluate the applicability of our BiSF method on other hESCs, two hESCs from WiCell (H1 and H9 cells) and one hESC established in Taiwan (hESC6) were recruited in this study. These three cell lines, grown in mTeSR1 media, were resuspended and formed embryoid bodies following the protocol in Figure 1A. All of these cells effectively differentiated into N-cadherin+ neural rosettes on D10 after treating with BiSF (Fig. 5A I, B I, C I). The ICC results revealed that most cells expressed the NPC markers SOX1 and PAX6 (Fig. 5A II, III, B II, III, C II, III). In addition to these epiblast-derived ESCs, a human fetal lung myofibroblast-derived iPSC (IMR90-1; from IMR-90 cells) and an amniotic fluid cell-derived iPSC (AF-iPSC) line were steered to NI under the BiSF condition. Both iPSCs showed efficient neural differentiation, evidenced by the extensive columnar neural rosettes displaying N-cadherin, SOX1, and PAX6 protein expressions (Fig. 5DI–III, E I–III). Notably, the BiSF method steers the neural differentiation of these tested pluripotent cells as efficient as that of TW1 hESCs. These data suggest that our BiSF method is applicable to both hESCs and iPSCs for efficient NI.
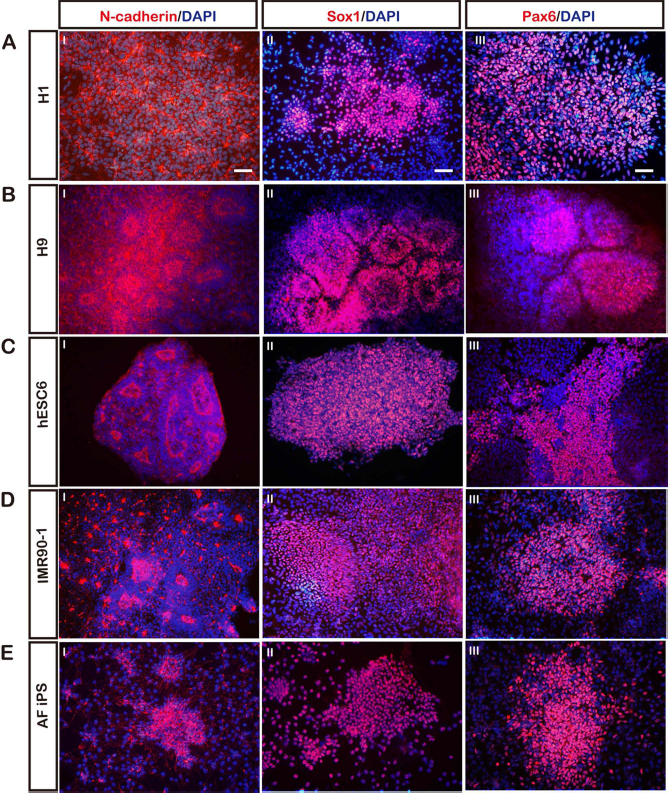
The NI efficacy of BiSF-treated hESCs and iPSCs. The NI efficacy of tested hESCs and induced pluripotent stem cells (iPSCs) was evaluated by the ICC staining for N-cadherin, SOX1, and PAX6 proteins. (A) H1 hESC. (B) H9 hESC. (C) hESC6. (D) IMR90-1 iPSC. (D) Amniotic fluid-derived (AF) iPSC. Scale bar: 50 μm.
Differentiating Potency of the Induced NPCs
Following the stage of NI in embryo, morphological patterning factors organize the neural tube with anterior– posterior (A-P) and dorsal–ventral (D-V) polarities and subsequently generate regionally restrictive neurons. For example, Dickkoph1 (DKK1), a Wnt antagonist secreted from the anterior visceral endoderm (AVE), polarizes NPCs to a forebrain neuron fate (10,30). Interestingly, we found that the transient treatment of the Wnt agonist BIO from D3 to D4 did not caudalize the default forebrain neuron property of the hESC-derived NPCs. The BiSF-induced NPCs were extensively marked with forebrainspecific antigens, including the BF-1 transcription factor (41) (89.7 ± 8.4%) (Fig. 6A) and Forse-1 membrane protein (40) (60.2 ± 7.3%) (Fig. 6B), but not posterior spinal cord markers, such as HOXB4 (33) (<1%) (Fig. 6C). In addition, the BiSF-induced NPCs were still competent to develop into spinal cord neural progenitors, as evidenced by the upregulated HOXB4 expression (50.7 ± 1.7%) after 0.5 μM RA treatment for 5 days (Fig. 6D).

BiSF-treated hESC progeny acquired an anterior CNS fate and the competency for regional patterning. (A, B) The NPCs derived from BiSF-treated TW1 cells showed the forebrain cell fate, characterized by the brain factor 1 (BF-1) and neural progenitor cell marker Forse-1 expression. (C) Homeobox B4 (HOXB4), a regional marker of the developing spinal cord, was not detected in the derived NPCs. (D) Treating the cells with 0.5 μM all-trans-retinoic acid (RA) from D10–D14 caudalized the NPCs and induced the expression of HOXB4. (E, F) The generation of dopaminergic neurons and spinal cord motor neurons was demonstrated by the tyrosine hydroxylase (TH) and the motor neuron and pancreas homeobox 1/homeobox HB9 (MNX1/HB9) transcription factor expressions, respectively. Scale bar: 50 μm.
These results suggest that the NPCs derived from BiSF-treated hESCs shared similar potency with the embryonic NPCs for responding to the A-P and D-V patterning factors. This speculation was further validated by showing that dopaminergic neurons (expressing tyrosine hydroxylase, 52.6 ± 2.2% of the cells) (Fig. 6E) and spinal motor neurons (HB9+ 53.2 ± 6.0% of the cells) (Fig. 6F) were effectively produced after receiving the FGF8/Shh (25) and RA/Shh signals (22), respectively.
BIO-Promoted OCT4 Expression Potentiates the NI of hESCs
BIO-activated canonical Wnt pathway has been shown to maintain hESC pluripotency and self-renewal (35). We also observed that for the early embryoid bodies of TW1 hESCs, providing BIO alone or BiSF prevented cell differentiation and sustained the pluripotent OCT4 protein expression, compared to the mock or SF treatment condition, respectively (Fig. 7). This result indicates that BIO-mediated self-renewal can be recapitulated in TW1 hESCs. However, these BIO properties of antidifferentiation and prorenewal in stem cells are inconsistent with the role of neural promotion in our BiSF method during hESC differentiation.
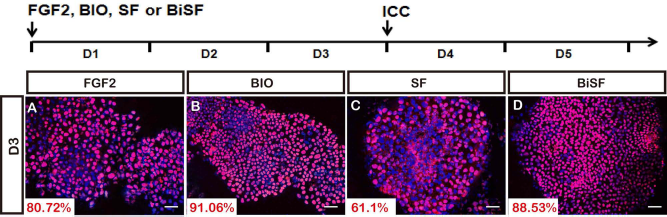
OCT4 protein expression in treated TW1 hESCs. The cells were treated with FGF (Mock) (A), BIO (B), SF (C), or BiSF (D) for 2 days. Their OCT4 expression was quantitatively evaluated by ICC staining. Scale bar: 50 μm.
To explore the underlying mechanisms, we first examined the BIO effect on the OCT4 expression of differentiating TW1 cells using ICC staining and quantitative RT-PCR analyses. Transient treatment of BiSF on D3 and D4 sustained a higher ratio of OCT4-expressing cells, compared to those in SF-treated hESCs (Fig. 8B I, II, C I, II, D I, II, E), although this comparison of OCT4 protein expression was not statistically significant. We further quantitatively evaluated the OCT4 mRNA expression in the treated hESCs using real-time RT-PCR. Compared to the SF condition at the NI stage, providing the BiSF treatment upregulated OCT4 mRNA expression on D3.5 (1.7-fold, p < 0.05), but oppositely abolished the OCT4 expression on D5 (0.17-fold, p < 0.01) (Fig. 8F). Interestingly, after removal of BIO and SB431542 on D5, a dramatic decline of OCT4 expression was observed in BiSF-treated hESCs (Fig. 8D III), but not in the FGF2 or SF-treated cells (Fig. 8B III, C III).

The OCT4 expression in FGF2-, SF-, and BiSF-treated TW1 hESCs. (A) The NI treatment and kinetic expression levels of OCT4 protein in the treated TW1 progeny were detected on D3, D4, and D5. (B–D) Immunostainings of Oct4 in FGF2 (B), SF (C), or BiSF (D) treated hESCs were revealed on D3, D4, and D5. (E) These ICC results were summarized and statistically analyzed. (F) OCT4 mRNA expression was quantified by the real-time RT-PCR. The data represented triplicate experimental results. Statistic results were analyzed by one-way ANOVA, and the significance was examined by Tukey's post hoc assay *p < 0.05. Scale bar: 50 μm.
Whether the upregulated OCT4 expression at the early stage of NI is required for the neural fate conversion of hESCs was further investigated. To test this speculation, endogenous OCT4 was modulated by specific OCT4-shRNA in the recombinant lentivirus-infected cells. The efficacy of the OCT4-shRNA lentivirus was validated by demonstrating a dramatically downregulated OCT4 protein expression in the infected TW1 cells (mock and SF-treated) on D3 at MOI 5 (p < 0.05) (Fig. 9A I–VI, B). Even in the presence of BIO, the OCT4-shRNA still reduced significant OCT4 expression in BiSF-treated cells on D4 (p < 0.05) (Fig. 9A VII–XI, B).
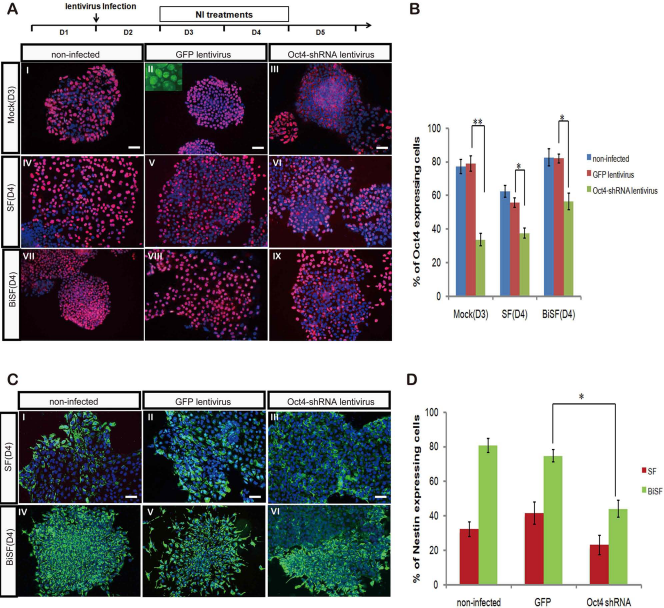
The NI efficacy in the TW1 hESCs infected with OCT4-shRNA lentivirus. (A) TW1 hESCs were infected with green fluorescent protein (GFP)- or OCT4-short hairpin RNA (shRNA) carrying lentivirus on D2 at a multiplicity of infection (MOI) of 5, and the levels of OCT4 protein expression were examined on D4. Viral infection and NI treatment schedules were illustrated. Inserted small panel in II shows the high efficacy of GFP lentivirus infection. (B) The inhibition efficacy of the OCT4-shRNA was evaluated and statistically analyzed. (C) After the lentiviral infection, the intracellular nestin expression was examined on D10 by ICC. (D) The nestin-expressing cells were quantified by flow cytometry analysis. *p < 0.05. Scale bar: 50 μm.
We next infected the hESCs with OCT4-shRNA lentiviruses on D2 and quantitatively determined the neural induction efficacy on D10 by flow cytometry analysis. The downregulated OCT4 expression in SF-treated hESCs led to a moderate NI reduction (Fig. 9C II, III), rather than NI promotion. Importantly, we demonstrate that OCT4-shRNA lentiviral infection (Fig. 9C VI), but not the control GFP lentivirus (Fig. 9C V), significantly reduced the ratio of nestin-expressing cells on D10 (p < 0.05) (Fig. 9D). These studies strongly demonstrate the prerequisite requirement of sustained OCT4 expression before neural fate conversion and an undiscovered role of Wnt activation for promoting NI of differentiating hESCs.
Discussion
Our study clearly demonstrates that treating human pluripotent stem cells with BiSF efficiently and rapidly steers the NI and produces the NPCs with forebrain neuron characteristics. These hESC-derived NPCs are competent for further differentiation by exogenous patterning factors and terminal maturation. All of the tested pluripotent cell lines, including H1, H9, TW1, hESC6, and AF-iPSCs, showed robust neural formation on D10 of differentiation. Notably, similar efficacy was achieved for the human fibroblast-derived IMR90-1 iPSCs, which was refractory to FGF2-triggered NI (16). This result suggests that the newly developed BiSF method is a potential general protocol to produce desired neuron precursors from hESCs and iPSCs. These findings on NI may improve the robust production and cell quality control for the neural cell transplantation of hESCs and iPSCs.
The differentiation of mESCs and hESCs has postulated to be an in vitro development model, recapitulating the embryonic developmental principles in lineage-determining processes and sequential cell fate specification (29). The FGF2-triggered NI of hESCs has demonstrated that PAX6 is initially and ubiquitously expressed in the NPCs before the expression of the SOX1 transcription factor (43). This in vitro finding was validated by the examinations in human embryos using RT-PCR and immunohistochemical staining.
Nevertheless, the expression priority of PAX6 in human neuroectoderm is inconsistent with that in mouse embryos (43). In our study, one interesting finding of the BiSF-induced NI is that SOX1 was almost uniformly detected in well-dispersed neural rosettes on D8 in hESC-derived NPCs. PAX6 expression was detected in less than 10% of cells on D8, but extensively expressed on D15. This observed expression profile of SOX1 and PAX6 is similar to that in the early developing neural tube of mouse embryos. Although the factors that contribute to the reversed expression sequence of SOX1 and PAX6 between mouse and human embryos are largely unknown, our studies imply that activated Wnt signaling in the neuroectoderm plays a major determinant role. We noticed that the SOX1 expression was initiated prior to PAX6 activation only when hESCs were cultured under BiSF conditions, but not under FGF2 or SF conditions. In addition, extending the treatment period of BiSF from D3 to D6 sustained SOX1 expression, but significantly abolished the induction of the PAX6+ cells (Fig. 3). Recent NI studies in mESCs demonstrate that SOX1 and PAX6 proteins are expressed in neuroepithelium and radial glia stages, respectively (39). Overexpressed SOX1 proteins sustain the multipotency of NPCs, but prevent their differentiation transition to radial glia and PAX6 expression (39). Based on these findings, we propose that the activated Wnt signals or the Wnt-triggered SOX1 enhancement may directly or indirectly contribute to the delayed PAX6 expression in the generated NPCs of the BiSF-treated hESCs.
PAX6 is a known neural determinant of hESCs (43). Both gain-of-function and loss-of-function assays support the essential role of PAX6 for the neural formation of hESCs (43). Although SOX1 was detected before PAX6 expression using the BiSF method, multipotent human NPCs were still obtained, and the NPCs can generate specialized neurons, such as midbrain dopaminergic neurons and spinal cord motoneurons. This result suggests that the prior expression of PAX6 is not a necessary condition for the generation of multipotent NPCs from hESCs. In addition, prolonged BiSF treatment from D3 to D6 significantly inhibited the PAX6 expression but still could produce nestin+/N-cadherin+/SOX1+ neural cells (Fig. 3B). Elucidating the characteristics of the SOX1+/PAX6- cells and comparing their intrinsic expression profiles to those of SOX1+/PAX6+ cells will reveal the detailed roles of PAX6 on the human NPCs. Examining the regulation of SOX1 and PAX6 may not only help the understanding of molecular evolution for controlling neural tube formation, but also the improvement of current methods for hESC neural differentiation.
Forebrain cell fate is postulated as the cell default property of the initial primitive neuroectoderm (29). The hESC-derived NPCs by FGF2 and/or SB431542 treatments are marked with forebrain characteristics and show the multipotent competence to form specific neurons (5,13,43). Canonical Wnt signal is a developmental patterning factor to caudalize anterior neurons and to specialize midbrain neuron formation (2). This Wnt signal in hESC-derived NPCs is antagonized by the endogenous DKK1 expression, and the attenuated Wnt activation consequently renders the establishment of anterior neuroectoderm bias (14). Treating Shh on D1 can inhibit the DKK1 and converts the anterior neuroectoderm fate to neural floor plate cells (14). Interestingly, adding BIO from D3 to D4 in our NI protocol did not interfere with the default forebrain cell fate of the generated NPCs, suggesting that the BIO-mediated Wnt activation was only transiently sustained during the NI stage, but did not last during the later neural specification stage. A similar finding was observed in a recent hESC neurogenic study using all-trans-RA (36), which is a caudalizing factor in developing neural tubes. Treating hESCs with RA in the presence of Noggin and SB431542 still can render uniformly forebrain NPCs and physiologically functional neurons (36). These results suggest that providing transient NI factors before D5 does not dramatically affect the later neural patterning and specification processes in the BiSF-treated hESCs.
Wnt signals are required for maintaining the self-renewal of stem cells and for preventing stem cell differentiation (28). Interestingly, our study provides in vitro evidence showing that BIO, a well-characterized glycogen synthase kinase (GSK)-3β inhibitor and a Wnt activator, promotes the neural differentiation of hESCs under the BiSF condition. Compared to the SB431542/FGF2 condition, adding BIO for 2 days led to transient OCT4 upregulation, but ultimately resulted in rapid OCT4 suppression. We postulate that the sustained OCT4 expression at the early NI stage (D3–D4) may prevent initiating nonneural lineage determinants, such as trophoectodermalor endomesodermal-specific genes. In addition, BIO treatments may prevent the reduction of the endogenous OCT4 level at the early NI stage and generate a synchronized hESC population (Fig. 7). These observations and postulations are supported by three recent studies in mESCs, revealing that sustained OCT4 expression promotes the NI specification, and knockdown of OCT4, in contrast, significantly attenuates the neural formation and lead to the trophoectodermal lineages (7,32,37). In addition, an efficient neural conversion of ESCs can be achieved by the coculture of bone marrow-derived PA6 cells (stromalderived neural induction activity, SDIA) (17). The SDIA is contributed, at least in part, by the pluripotent maintenance capability of PA6 cells on differentiating mESCs (7,37). These findings strongly emphasize the indispensible role of pluripotent maintenance before the neural commitment of hESCs.
In differentiating hESCs, shutdown of pluripotent machinery is a prerequisite to priming cell differentiation and lineage-specific genes (15). The efficient neural formation using BiSF was accompanied by a rapid decline of OCT4 expression on late NI stage (D5), compared to the SF-treated hESCs. One recent report indicates that inhibition of activin/nodal downstream mothers against decapentaplegic 2 (SMAD2) activity accelerates the decline of OCT4 and Nanog expressions but sustains the SOX2 level in the FGF2/Noggin-treated hESCs (15). Whether BIO acts cooperatively with SB431542 to inactivate the SMAD2 activity will be further explored. Understanding the timed control of downregulation of pluripotent genes in the BiSF-treated hESCs requires detailed examinations of the signal networks of SMADs, GSK-3β, and extracellular signal-regulated kinase (ERK) pathways. Further investigations on the NI of hESCs using the BiSF method will provide more details of human neural tube formation and the cellular intrinsic controlling machinery regulated by wired BMP, Wnt, and FGF signals.
Conclusion
Here an efficient NI method using BiSF treatment for hESCs and iPSCs is introduced. The hESC-derived NPCs exhibit forebrain cell fate and a prior expression order of SOX1 before PAX6, similar to the pattern in the early developing neural tube of mouse embryo. Transient Wnt activation during the NI does not caudalize the anterior cell polarity and abrogate the competency for responding to later neural patterning factors. We further demonstrate that the Wnt signal prevents the downregulation of OCT4 expression in differentiating hESCs, and the sustained OCT4 level on the initial stage of NI is essential for the commitment of neuroectoderm.
Footnotes
Acknowledgments
This work was supported by grants from the National Science Council of Taiwan (NSC 99-2628-B-005-015-MY3 and NSC 102-2628-B-005-007-MY3) and the Taichung Veterans General Hospital/National Chung Hsing University Joint Research Program (TCVGH-NCHU 1017604 and 1027607). This research was also funded in part by the Ministry of Education, Taiwan, Republic of China, under the ATU plan. Assistances provided by Dr. Fu-Chou Cheng and Dr. Shiaw-min Huang, at the Taichung Veterans General Hospital and the Food Industry Research and Development Institute, respectively, are highly appreciated. The authors declare no conflicts of interest.