
Abstract
Even after decades of intensive studies, therapeutic options for patients with stroke are rather limited. Thrombolytic drugs effectively treat the very acute stage of stroke, and several neuroprotectants that are designed to treat secondary injury following stroke are being tested in clinical trials. However, these pharmacological approaches primarily focus on acute stroke recovery, and few options are available for treating chronic stroke patients. In recent years, stem cell-mediated regenerative approaches have emerged as promising therapeutic strategies for treating the chronic stage of stroke. In this study, we examined whether systemically administered bone marrow cells (BMCs) could have beneficial effects in a rat model of chronic ischemia. Our transplantation experiments using BMCs obtained from ischemic donor rats showed functional and structural recovery during the chronic stage of stroke. BMC-mediated neural proliferation was prominent in the brains of rats with chronic stroke, and most of the new cells eventually became neurons instead of astrocytes. BMC-mediated enhanced neural proliferation coincided with a significant reduction (~50%) in the number of activated microglia, which is consistent with previous reports of enhanced neural proliferation being linked to microglial inactivation. Strikingly, approximately 57% of the BMCs that infiltrated the chronic ischemic brain were CD25+ cells, suggesting that these cells may exert the beneficial effects associated with BMC transplantation. Based on the reported anti-inflammatory role of CD25+ regulatory T-cells in acute experimental stroke, we propose a working model delineating the positive effects of BMC transplantation during the chronic phase of stroke; infiltrating BMCs (mostly CD25+ cells) reduce activated microglia, which leads to enhanced neural proliferation and enhanced recovery from neuronal damage in this rat model of chronic stroke. This study provides valuable insights into the effect of BMC transplantation in the chronic ischemic brain, which may lead to the development of effective therapy for chronic stroke patients who currently lack satisfactory therapeutic options.
Introduction
Stroke is the leading cause of disability and a primary cause of death in developed countries (2,30). Treatment with thrombolytic drugs is successful if the drugs are administered within 3–4.5 h of the onset of symptoms (13,32); however, this short treatment window allows less than 5% of acute stroke victims to receive this therapy. Unfortunately, with the exception of thrombolytic therapy, no proven treatment for stroke has been demonstrated despite extensive efforts during the past few decades.
In recent years, cell-based therapy has emerged as a promising new strategy to treat stroke patients. Various types of stem cells, including embryonic stem cells (ESCs) (7), neural stem cells (NSCs) (19), and bone marrow stromal cells (BMSCs) (8), have been tested for their efficacy in animal stroke models. However, ESC-derived cell populations retain tumorigenic potential; NSCs cannot be easily obtained from patients; and the number of BMSCs that can be extracted from a simple biopsy is rather limited in quantity, necessitating ex vivo expansion.
Bone marrow cells (BMCs) have emerged as a promising cell source for autologous cell-based therapy, especially in emergent cases, because these cells can be directly used for transplantation without ex vivo culturing. BMCs are a heterogeneous cell population consisting of hematopoietic stem cells, BMSCs, and various immune cells, including T-cells, B-cells, and monocytes (7). Additionally, many cell types among the BMC population secrete various therapeutically beneficial cytokines (31).
The therapeutic efficacy of BMC transplantation was initially tested in the treatment of myocardial infarction both in animals (24) and human patients (25). In subsequent studies, BMC administration was shown to promote functional recovery during the acute stage of cerebral infarction in rat models of stroke (6,9,12,18). Furthermore, the administration of BMCs to acute stroke patients was shown to enhance functional recovery (26).
Most studies about the efficacy of BMCs have focused on the acute phase of stroke, and so the question of whether BMC transplantation provides beneficial effects in chronic stroke patients requires further investigation. A recent study addressing the biodistribution of intra-arterially (IA) injected BMCs demonstrated that the BMCs migrated to the brains of chronic stroke patients; this result suggests that BMCs may exhibit therapeutic benefits even during the chronic phase of stroke (5). However, up until now, none of the therapeutic effects of BMC transplantation during the chronic phase of stroke has been reported.
Here, we demonstrate that BMC transplantation during the chronic stage of ischemic stroke exerts positive effects on brain structure and behavioral performance using a rat model of focal ischemic stroke. The results of this study provide valuable insight into the development of cell-based therapeutic approaches for the treatment of patients suffering from chronic ischemic stroke.
Materials and Methods
Animal Model
CHA University Institutional Animal Care and Use Committee approved this study. Middle cerebral artery occlusion (MCAO) was used to generate a rat model of ischemic stroke. The surgery was conducted in adult male Sprague–Dawley rats (n = 30 per group) (Orient Bio, Seongnam, Korea) weighing 240 to 270 g. They were anesthetized with 5% isoflurane (Hana Pharm, Hwasung, Korea) and maintained with 2% isoflurane in a mixture of 70% N2 and 30% O2. The common carotid artery, internal carotid artery (ICA), and external carotid artery (ECA) were exposed by a cervical incision, and the pterygopalatine and occipital arteries were then cauterized. Blood flow was temporally blocked by ligating the ICA and ECA. A 23.0-mm-long 4–0 nylon monofilament (Ailee, Busan, Korea), coated with low viscous silicone (Handae Chemicals, Chungbuk, Korea), was advanced from the exposed ECA to the ICA. The monofilament occluded the middle cerebral artery. At a total of 1.5 h after MCAO, reperfusion was induced by withdrawing the suture from the ECA.
BMC Preparation
For transplantation, allogeneic BMCs were obtained from the time-matched donor ischemic rats at 5, 10, 11, and 12 weeks after the MCAO surgery. The femur, tibia, and fibula were removed from the donor rats and homogenized in 30 ml of 5 mM ethylenediaminetetraacetic acid (EDTA)/0.1 M phosphate-buffered saline (PBS) (Welgene, Daegu, Korea) using a mortar and pestle. BMCs were filtered through a 100-μm cell strainer (BD Biosciences, Franklin Lakes, NJ, USA) and then treated with red blood cell (RBC) lysis buffer (Qiagen, Valencia, CA, USA) for 5 min at 37°C to remove the erythrocytes. BMCs were injected into animals within 10 min after preparation.
For immunophenotypic analysis of the composition of the BMC fraction, BMCs were obtained from sham control rats at 1 week postsurgery and ischemic rats at either 1 week or 5 weeks after MCAO induction using the method described above.
Transplantation of BMCs
Allogeneic BMCs were injected into the MCAO rats both intra-arterially and intravenously. Briefly, the rats were anesthetized with a mixture of 2% isoflurane in 70%/30% nitrogen/oxygen. For the intra-arterial (IA) injection, we exposed the ECA, punctured a small opening about 2–3 mm below the ligation site in the ECA, inserted PE10 polyethylene tubing (BD Biosciences) through the puncture in the ECA up to the ICA, and injected 2 × 107 cells in 1 ml of 0.1 M PBS using a 1-ml syringe (BD Biosciences) that was equipped with a 28-gauge needle (BD Biosciences) that was attached to the other end of the tubing. After the cells were slowly injected over 2 min, the tubing was withdrawn, and the hole was cauterized to avoid hemorrhage.
An intravenous (IV) injection was administered through the tail vein using a 1-ml syringe (BD Biosciences) that was equipped with a 28-gauge needle (BD Biosciences). Approximately 2 × 107 cells in 1 ml of 0.1 M PBS were injected at a time per rat. Control rats were injected with 1 ml of 0.1 M PBS.
Labeling and Tracking of BMCs
A total of 2 × 107 BMCs in 1 ml of 0.1 M PBS were labeled with 25 μM of a fluorescent dye, carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) (Invitrogen, Carlsbad, CA, USA), at 37°C for 10 min. The labeled cells were injected into sham and chronic ischemic rats both IA and IV (2 × 107 BMCs in 1 ml of 0.1 M PBS per injection) as described above. We used this cell dose (2 × 107 cells in 1 ml of PBS) based on the previous reports that systemic treatment with 1~3 × 107 cells was effective in promoting recovery (9,11,12,17,18,34).
Behavioral Tests
The modified neurologic severity score (mNSS) and foot fault tests were performed 1 day before the MCAO surgery and every week for 13 weeks after the MCAO surgery. The mNSS was used to assess motor (muscle status and abnormal movement), sensory (visual, tactile, and proprioceptive), and reflex (pinna, corneal, and startle) functions. The test results were indexed as a grade that ranged from 0 to 18 (0, no impairment; 1–6, mild impairment; 7–12, moderate impairment; and 13–18, severe impairment).
The foot fault test was performed by placing rats on a homemade wire grate (50 cm × 50 cm with 2.5 cm × 2.5 cm grids) for 5 min and was recorded by videotaping from below the grate. The number of forelimb slips through the grate was counted during the 5-min test session and was converted into the foot fault score as follows: foot fault score = (number of faults of the affected forelimb/number of footsteps of the affected forelimb) – (number of faults of the nonaffected forelimb/number of footsteps of the nonaffected forelimb).
Flow Cytometry Analysis
The following antibodies were used to analyze the cell types that composed the BMC population: fluorescein isothiocyanate (FITC)-conjugated mouse monoclonal anticluster of differentiation 4 (CD4; Biolegend, San Diego, CA, USA), mouse monoclonal anti-CD8 (Biolegend), mouse monoclonal anti-CD25 (Thermo Scientific, Inc., Bremen, Germany), mouse monoclonal anti-CD11b/c (Abcam, Cambridge, UK), rabbit polyclonal anti-CD20 (BD Biosciences), monoclonal anti-CD31 (BD Biosciences), chicken polyclonal anti-CD294 (Sigma-Aldrich, St. Louis, MO, USA), goat polyclonal anti-T-cell immunoglobulin mucin family member 3 (TIM-3; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), Alexa 647-conjugated hamster monoclonal anti-CD29 (Biolegend), and mouse monoclonal anti-CD45 (Biolegend). All antibodies were used at 5 μg/ml. The BD FACSCalibur Flow Cytometry System (BD Biosciences) was used for the analysis.
5-Bromo-2-Deoxyuridine (BrdU) Labeling
BrdU (50 mg/kg in saline; Sigma-Aldrich) was administered daily for 2 weeks by an intraperitoneal (IP) route starting from 2 weeks after transplantation of BMCs; we injected BrdU from 2 weeks posttransplantation to take into account the time taken for the BMCs administered into the bloodstream to be able to confer any effect in the brain. The rats were anesthetized using ketamine (50 mg/kg, IP; Huons, Seoul, Korea) and xylazine (5 mg/kg, IP; Bayer Korea, Seoul, Korea). The animals were perfused by transcardiac perfusion using 0.1 M PBS, which was followed by 4% formaldehyde (Samchun Chemicals, Pyeong Taek, Korea). The brains were removed and fixed in 4% formaldehyde; they were cryoprotected sequentially in 15% and 30% sucrose (Sigma-Aldrich) and frozen at −80°C in an optimal cutting temperature (OCT) solution (Sakura Finetek, Torrance, CA, USA). The brain sections were cut at a thickness of 30 μm using a cryostat (Microm, HM 525, Walldorf, Germany) and then stored in antifreezing buffer [30% glycerol (Samchun Chemicals)/30% ethylene glycol (Junsei Chemical Co., Tokyo, Japan) in 0.1 M PBS]. To perform BrdU staining, the brain sections were incubated in 2 N HCl (Samchun Chemicals) at 37°C for 30 min and neutralized in 0.1 M sodium borate (pH 8.0; Sigma-Aldrich) for 20 min. The sections were then incubated in blocking solution [10% goat serum (Vector Laboratories, Burlingame, CA, USA)/0.1% NP-40 (Sigma-Aldrich) in 1× Tris-buffered saline (TBS; Sigma-Aldrich)] for 1 h, which was followed by incubation with 2 μg/ml of a rat monoclonal anti-BrdU antibody (Abcam) at 4°C for 12 h. The sections were then washed three times in 1× TBS, incubated with 2 μg/ml of a goat Alexa 594-conjugated anti-rat IgG antibody (Invitrogen), and then examined with a LSM510 confocal microscope (Carl Zeiss, Göttingen, Germany).
Administration of Minocycline
Minocycline (40 mg/kg in saline, IP; Sigma-Aldrich) was injected into ischemic rats for 4 consecutive days starting from 4 weeks post-MCAO surgery, and animals were sacrificed at 1 week after the last injection of the drug.
Immunohistochemistry
Immunostaining was performed as described above. The following primary antibodies were used: rabbit polyclonal anti-doublecortin (anti-DCX, 2 μg/ml; Cell Signaling Technology, Danvers, MA, USA), mouse monoclonal anti-polysialic acid-neural cell adhesion molecule (anti-PSA-NCAM, 2 μg/ml; Millipore, Billerica, MA, USA), mouse anti-glial fibrillary acid protein (anti-GFAP, 2 μg/ml; Millipore), mouse anti-neuronal nuclei (NeuN; 2 μg/ml; Millipore), rabbit polyclonal anti-ionized calcium-binding adapter molecule 1 (anti-Iba1, 2 μg/ml; Wako Pure Chemical Industries, Osaka, Japan), and mouse monoclonal anti-macrophages/monocytes clone ED1 [anti-ED1 (CD68), 2 μg/ml] (Millipore). The following secondary antibodies (4 μg/ml) were used in this study: goat Alexa 594-conjugated anti-rat IgG, goat Alexa 594-conjugated anti-mouse IgG, goat Alexa 594-conjugated anti-mouse IgM, goat Alexa 594-conjugated anti-rabbit IgG, goat Alexa 633-conjugated anti-mouse IgG, and goat Alexa 633-conjugated anti-rabbit IgG (Invitrogen). Hoechst dye (bisBenzimide H33342, 1 μg/ml; Sigma-Aldrich) was used for labeling nucleic acids. The sections were examined with an LSM510 confocal microscope (Carl Zeiss).
Measurement of Infarction Volume
Three 30-μm-thick coronal sections at the levels of 1.7 mm, 0.7 mm, −0.3 mm, −1.3 mm, −2.3 mm, and −3.3 mm from bregma were cut and mounted on silanecoated glass slides (Marienfeld, Lauda-Koenig-Shofen, Germany). The sections were stained with 0.2% cresyl violet (Nissl; Sigma-Aldrich) and imaged using a dissecting microscope that was equipped with a digital camera (Olympus, Tokyo, Japan). The size of the infarct area was measured using the NIH ImageJ software program (National Institutes of Health, Bethesda, MD, USA). The reduced brain volume was calculated as follows: brain volume reduction = (size of the infarct area in the ipsilateral hemisphere)/(size of the contralateral noninfarct hemisphere).
Statistical Analyses
The data are presented as the mean ± SEM. The behavioral scores for the mNSS and foot fault test were evaluated using an ANOVA analysis with repeated measures. Differences between the control and BMC-injected groups were assessed using Student's t test or by a one-way ANOVA that was followed by the Student–Newman–Keuls post hoc test. Differences were considered to be statistically significant at p < 0.05.
Results
Characterization of Bone Marrow Cells
BMCs are a heterogeneous population of different cell types, so we examined the composition of the BMC fraction using an immunophenotypic analysis. In this analysis, BMCs were obtained from sham control rats at 1 week postsurgery and ischemic rats at either 1 week or 5 weeks after MCAO induction. Our results demonstrated that the BMC composition from the ischemic rats was different from that of the time-matched (e.g., 1 week after surgery) sham control rats, especially regarding the percentages of CD4+ cells (helper T-cells) and CD25+ cells (regulatory T-cells and activated B-cells) (Fig. 1A, B). In addition, the cell composition of the BMCs that were obtained 1 week after the MCAO surgery was different from that of the BMCs that were obtained 5 weeks after the ischemic surgery. For example, the percentages of CD4+ cells (helper T-cells), CD8+ (cytotoxic T-cells), CD11b+/c+ (macrophages, granulocytes, and dendritic cells), and TIM-3+ cells (type 1 helper T-cells) were significantly higher at 5 weeks than at 1 week postischemia (Fig. 1A, C–E). In contrast, the percentages of CD31+ cells (endothelial cells, macrophages, and neutrophils), CD294+ cells (type 2 helper T-cells), CD20+ cells (B-cells), CD45+ (hematopoietic cells), CD29+ (leukocytes, granulocytes, and mesenchymal stem cells), and CD29+/CD45- cells (mostly bone marrow mesenchymal stem cells) (1,14,15) were not significantly altered by ischemic injury or at different time points after the injury (Fig. 1F–K).
Immunophenotypic characterization of BMCs. To examine the compositions of BMC populations obtained from various conditions, BMCs were prepared from control rats 1 week after sham surgery and 1 and 5 weeks after MCAO induction. The prepared BMCs were labeled with the following surface markers and analyzed according to the percentage of each cell type by fluorescent-activated cell sorting (FACS) analysis: (A) cluster of differentiation 4 (CD4), a marker for pan helper T-cells; (B) CD25, a marker for regulatory and activated T-cells and activated B-cells; (C) CD8, a marker for cytotoxic T-cells; (D) CD11b/c, a marker for macrophages, granulocytes, and dendrocytes; (E) T-cell immunoglobulin mucin family member (TIM-3), a marker for type 1 helper T-cells; (F) CD31, a marker for endothelial cells, macrophages, and neutrophils; (G) CD294, a marker for type 2 helper T-cells; (H) CD20, a marker for B-cells; (I) CD45, a marker for hematopoietic cells; (J) CD29, a marker for mesenchymal stem cells (MSCs), leukocytes, and granulocytes; and (K) CD29+ CD45-, a marker for bone-marrow MSCs. The statistical analysis was performed using a one-way ANOVA, which was followed by a Student–Newman–Keuls post hoc test to compare the groups. N = 3/group, *p < 0.05, **p < 0.01.
Altogether, these results indicate that the percentages of certain cell types within the BMC population change during ischemic stroke and at different time points following the onset of ischemia.
Cell Tracking During the Chronic Stage of Ischemic Stroke
To estimate the number of BMCs that infiltrated the brain during the chronic phase of ischemic stroke, we injected CFDA-SE-labeled BMCs both IA and IV (2 × 107 cells per injection) into rats at 4 weeks after the sham and MCAO surgeries were performed. The BMCs used for CFDA-SE labeling were obtained from time-matched chronic ischemic rats (4 weeks post-MCAO surgery). Each group of rats was sacrificed 3 h, 1 day, or 7 days after the injection, and the number of CFDA-SE+ cells in the brain was counted (Fig. 2A). Three hours after the cell injection, the number of labeled cells increased significantly in the ipsilateral hemisphere of the MCAO rats compared to the sham control group (197 vs. 34) (Fig. 2B). Intriguingly, the number of labeled BMCs was dramatically reduced down to 18 at 1 day after the cell injection, which is consistent with the observation that was made in a previous report using a rat model of acute stroke (6). However, we did not observe further reduction of the infiltrated cells for at least 7 days afterward. Therefore, about 9% of the infiltrated cells detected in the ipsilateral ischemic brain at 3 h postinjection persisted in the brain for at least 7 days.
Brain infiltration of BMCs. Carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) was used to track the transplanted BMCs. The CFDA-SE-labeled BMCs were injected into the sham and MCAO rats (via both IA and IV), and the distribution of the transplanted BMCs in the rat brain was tracked. (A) Left panels, CFDA-SE-labeled cells; middle panels, Hoechst 33342-labeled cells; right panels, merged image. Scale bar: 10 μm. (B) Total numbers of CFDA-SE+ cells in the ipsilateral hemisphere of the sham and MCAO rats after BMC transplantation at 4 weeks postsurgery. The sham group was sacrificed for analysis 3 h posttransplantation, and the MCAO group was sacrificed at three different time points (3 h, 1 day, and 7 days) after transplantation. The number of BMCs that infiltrated into the brain decreased dramatically between 3 h and 1 day after BMC transplantation. N = 3/group, *p < 0.05, w, weeks.
Our results indicate that BMC infiltration was increased by ischemic injury even in the chronic phase of stroke because the number of infiltrating BMCs was significantly higher in the ischemic brain than in the sham control.
BMC Transplantation Promotes Behavioral Recovery in Chronic Ischemic Rats
To examine the effects of allogeneic BMCs that were delivered during the chronic stage of stroke, we injected the cells at 5 weeks postischemia both IA and IV (2 × 107 cells per injection). In addition, we injected BMCs at 10, 11, and 12 weeks postischemia via only noninvasive IV administration to enhance the efficacy of BMC transplantation. The transplanted allogeneic BMCs were prepared from donor rats in which ischemic injury was induced at the same time as the recipient rats (i.e., the BMCs from the donor ischemic rats 5, 10, 11, and 12 weeks postischemic injury were transplanted into the stroke rats that were also 5 10, 11, and 12 weeks postischemia, respectively). The behavioral performance was evaluated almost every week during the entire experimental period (Fig. 3A).
Functional recovery after the transplantation of BMCs. BMCs were transplanted into the chronic stroke rat model, and their effect on behavioral performance was examined. (A) Schedule of the experiment. BrdU, 5-bromo-2-deoxyuridine. The behavioral performances of the rats in each group were measured with the mNSS (B) and the foot fault test (C). N = 10 for the control group, N = 13 for the BMC-injected group. **p < 0.01, *p < 0.05, the data are the means ± SEM.
mNSS was not significantly improved by the MCAO transplantation until 6 weeks after the first BMC injection (i.e., +11 weeks). However, the mNSS score of the BMC-treated group began to display a statistically significant difference compared with the control rats at 7 and 8 weeks (i.e., +12 and 13 weeks) posttransplantation (Fig. 3B). In contrast, the foot fault test, which is a sensitive behavioral test (27), showed a significant functional improvement as early as the first week after the BMC transplantation, that is, +6 weeks (Fig. 3C).
Altogether, our results indicate that BMC transplantation improves functional recovery even in the chronic stage of ischemic stroke.
BMC Transplantation During the Chronic Stage of Stroke Promotes Recovery From Brain Tissue Damage
Behavioral improvement was evident after the BMC transplantation, so we examined whether BMC delivery promoted tissue recovery from ischemia-mediated brain damage. Nissl staining (cresyl violet) of the ischemic brain sections demonstrated that the BMC treatment reduced the damaged brain area when compared with vehicle (0.1 M PBS)-treated controls [43% in the BMC group (n = 14) vs. 55% in the control group (n = 9), p < 0.05] (Fig. 4).
Enhanced brain structural recovery after BMC transplantation. (A) Nissl staining was performed to measure the area of brain damage. The numbers on the upper right corner of each panel indicate the level of each brain section with reference to bregma. (B) The percentage of the damaged area in the ipsilateral hemisphere with regard to the contralateral hemisphere was compared between the vehicle and BMC-injected groups. The rats were sacrificed and analyzed 8 weeks after the BMC transplantation. Seven sections that were separated 1 mm apart from each other were analyzed for the percentage of remaining intact ipsilateral areas compared with the contralateral hemisphere. N = 9 for the control group, N = 14 for the BMC-treated group. *p < 0.05.
We speculated that this reduced brain damage by BMC transplantation may at least partly be caused by induction of neural proliferation as seen in the acute stage of ischemic stroke (3,20). To examine whether the transplanted BMCs promoted neural proliferation in chronic ischemic rats, we injected BMCs (via both IA and IV) at 5 weeks after the MCAO surgery. The animals were subsequently sacrificed, and an immunohistological analysis was performed 8 weeks after surgery. To mark the new cells, BrdU was administered daily for 2 weeks starting on the second week after the first BMC transplantation. We observed a dramatic increase (approximately threefold) in the number of neural precursor cells that expressed DCX and PSA-NCAM in the subventricular zone (SVZ) (p < 0.05, n = 10 for the control, n = 8 for the BMC group) (Fig. 5A). We also detected a significant increase (approximately 2.6-fold) in the number of new neurons (BrdU+ and NeuN+) near the peri-infarct area upon BMC transplantation (Fig. 5B). Conversely, no BrdU-labeled astrocytes (BrdU+ and GFAP+) were detected in the peri-infarct region (Fig. 5C). This result indicates that the BMC transplantation during the chronic stage of stroke also promoted neural proliferation in the SVZ, and most of the newly generated cells near the peri-infarct area were neurons.
Increased neural proliferation in the SVZ after BMC transplantation. (A) The expression level of two neuroblast markers, doublecortin (DCX) and polysialic acid-neural cell adhesion molecule (PSA-NCAM), in the SVZ was compared between the control and BMC groups. The percentage of PSA-NCAM-positive area per high-power field was measured because of the difficulty in counting the number of individual cells. N = 10 for the control group, N = 8 for the BMC group. *p < 0.05, scale bar: 10 μm. (B) Costaining of neuronal nuclei (NeuN, a neuronal marker) and BrdU (a marker for new cells) in the peri-infarct area indicated that most of the new cells became neurons in this area. In addition, BMC transplantation dramatically enhanced the number of BrdU+ cells in comparison with the vehicle (0.1 M PBS) injection. N = 4 for the control group, N = 5 for the BMC group. *p < 0.05, scale bar: 10 μm. (C) In contrast, new cells (BrdU+) rarely express an astroglial marker, glial fibrillary acidic protein (GFAP), in the peri-infarct area in both the vehicle and BMC-treated groups. N = 4 for the control group. N = 5 for the BMC group. *p < 0.05, scale bar: 10 μm.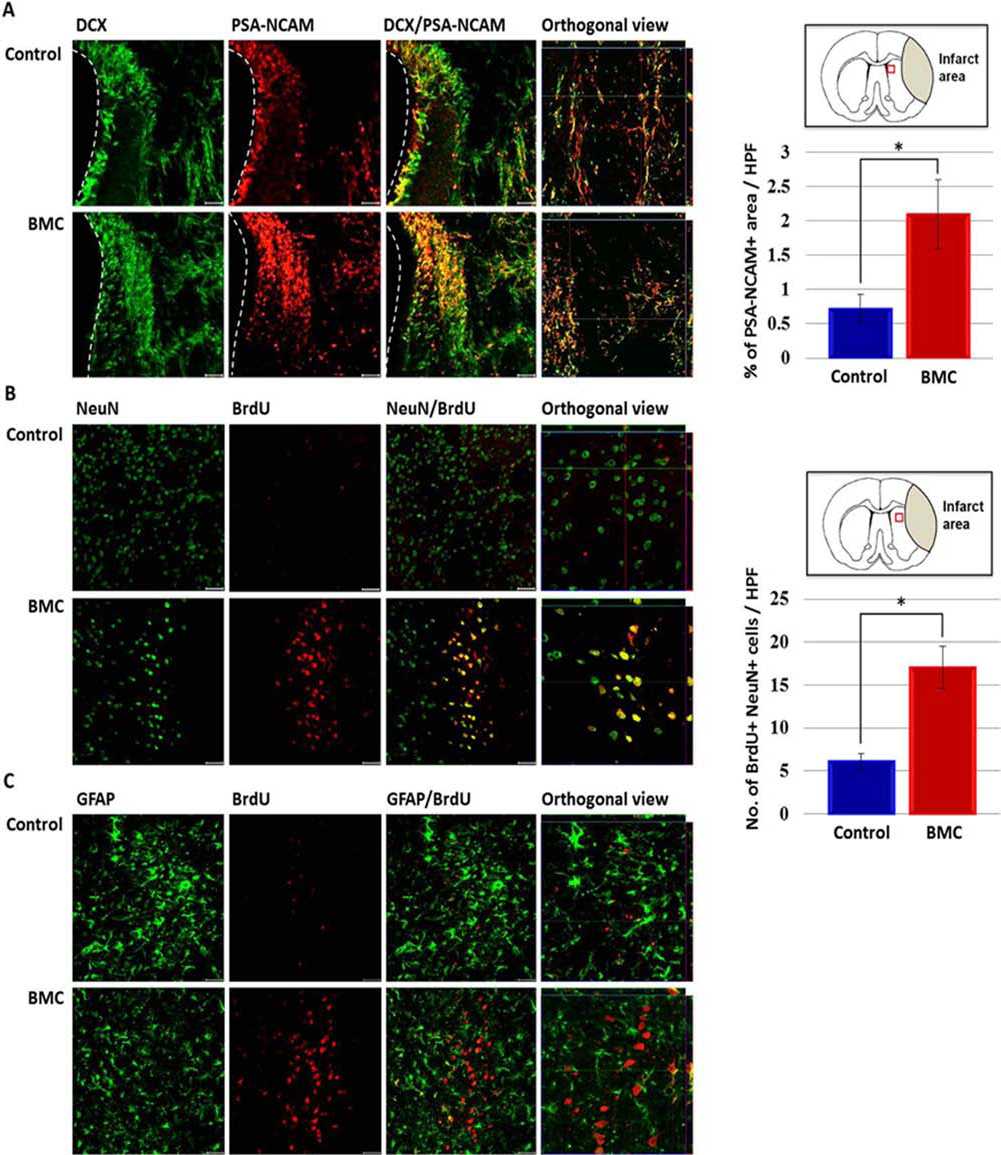
We next examined whether the increased neural proliferation from the BMC transplantation also occurred in the dentate gyrus, which is another prominent neurogenic region in the brain. As shown in Figure 6A, an increase (approximately twofold) in the expression of DCX and PSA-NCAM in the dentate gyrus of the BMC-treated rats was observed. Similar to the SVZ, the number of new cells (BrdU+ cells) was also much higher in the hippocampus of the BMC-treated group than in the vehicletreated group, and most of the new cells were neurons (NeuN+) and not astrocytes (GFAP+) (Fig. 6B, C).
Increased neural proliferation in the dentate gyrus after BMC transplantation. (A) The number of cells expressing DCX and PSA-NCAM, two neuroblast markers, in the dentate gyrus were compared between the control and BMC groups. N = 10 for the control group, N = 8 for the BMC group. *p < 0.05, scale bar: 10 μm. (B) Costaining of NeuN (neuronal marker) and BrdU (marker for new cells) in the peri-infarct area indicated that most of the new cells became neurons in this area. In addition, BMC transplantation dramatically enhanced the number of BrdU+ cells compared with the vehicle (0.1 M PBS) injection. N = 4 for the control group. N = 5 for the BMC group. *p < 0.05, scale bar: 10 μm. (C) In contrast, new cells (BrdU+) rarely express the astroglial marker, GFAP, in the peri-infarct area in both the vehicle and BMC-treated groups. N = 4 for the control group. N = 5 for the BMC group. *p < 0.05, scale bar: 10 μm.
Taken together, our results consistently demonstrate that BMC transplantation during the chronic stage of stroke promotes neural proliferation in the ischemic brain. This response occurred both in the SVZ and the dentate gyrus, and most of these new cells eventually differentiated into neurons.
Decreased Microglial Activation After BMC Transplantation
It has been suggested that the reduced phagocytic activation of microglia promotes neural proliferation (4,33). Therefore, we examined whether a reduction in microglial activation was observed after BMC transplantation. When the brain sections were immunostained for both Iba1 (a general marker for microglia, although upregulated in activated microglia) and ED1 (a marker specific for activated microglia), an approximately 46% reduction in the number of ED1+ cells near the peri-infarct striatum of the BMC-treated group was observed when compared with the vehicle-injected rats. The number of Iba1+ cells was also reduced by transplantation of BMCs, although not as dramatically as that of ED1+ cells (Fig. 7A). This result clearly indicates that activated microglia still existed in the striatum of the chronic ischemic rats. Moreover, BMC administration may confer a beneficial effect by at least partially suppressing the number of activated microglial cells (Fig. 7A). A similar observation was also observed in the dentate gyrus of the hippocampus (Fig. 7B).
Reduced microglial activation after BMC transplantation. (A) The number of activated microglia (ED1+ cells) at the chronic stage of stroke was significantly reduced after BMC transplantation in the striatum. The rightmost panels of the pictures are magnified images of the white boxes shown in panels on the left side. *p < 0.05, scale bar: 10 μm. Iba1, ionized calcium-binding adapter molecule. (B) The percentage of activated microglia (ED1+Iba1+) among total microglia (Ib1+) in the dentate gyrus. N = 9/group. *p < 0.05, scale bar: 10 μm.
Taken together, the microglial activation that was still occurring in the chronic ischemic brain was reduced by up to 50% after BMC transplantation, and this effect may, at least in part, contribute to increased neural proliferation in the transplanted brain.
Reduced Microglial Activation and Increased Neural Proliferation by Administration of Minocycline
To support the link between microglial inactivation and enhanced neural proliferation during the chronic ischemic phase, we sought to suppress microglial activation by administration of minocycline (Fig. 8A). First of all, we detected a significant reduction in the damaged brain area when minocycline was injected at 4 weeks after MCAO surgery (Fig. 8B). As expected, the number of activated microglia (ED1+ cells) was significantly reduced (Fig. 8C), while the number of BrdU+ cells was greatly increased (Fig. 8D), after the minocycline administration. A significant portion of the BrdU+ cells were colabeled with PSA-NCAM antibody, suggesting that the BrdU+ cells were derived from the proliferation of neural stem cells in the SVZ area. The rest of the BrdU+ cells that did not express PSA-NCAM might be either due to differentiation of the neural cells or they originate from outside the brain. In contrast, administration of minocycline into normal rats did not change the number of BrdU+ cells and Iba1+ cells, indicating that minocycline per se had no direct effect on neural proliferation (Fig. 8E, F).
Reduced microglial activation and increased neural cell proliferation by minocycline. (A) Experimental schedule. Both minocycline (40 mg/kg in saline, IP; Sigma-Aldrich) and BrdU (50 mg/kg in saline, IP) were injected into ischemic rats for 4 days daily starting from 4 weeks post-MCAO surgery, and animals were sacrificed at 1 week after the last injection of the minocycline. (B) Enhanced brain structural recovery by minocycline administration. (C) Reduction of activated microglia in the striatum by the administration of minocycline. (D) Neural cell proliferation was greatly increased in the striatum by the administration of minocycline. The rightmost panels of the pictures are magnified images of white boxes shown in panels on the left side. (E) The number of activated microglia in the striatum of normal rats was not changed by the administration of minocycline. (F) The number of newborn cells in the striatum of normal rats was not changed by the administration of minocycline. N = 5/control group, N = 6/minocycline-treated group. *p < 0.05, scale bar: 10 μm.
Taken together, our results demonstrated that administration of minocycline reduced activated microglia and increased neural cell proliferation in the chronic ischemic brain. This result supported our hypothesis that reduction of activated microglia by BMC transplantation contributed at least partly to the neural cell proliferation in the chronic ischemic brain.
CD25+ Cells Infiltrated the Ischemic Brain
One plausible explanation that links BMC transplantation to a reduction in microglial activation is the infiltration of CD25+ cells (i.e., regulatory T-cells) into the ischemic brain. It has been reported that CD25+ regulatory T-cells reduce microglial activation by secreting anti-inflammatory cytokines (21). Therefore, we examined whether the CD25+ cells that exist within the BMC population infiltrated the ischemic hemisphere. When we transplanted CFDA-SE-labeled BMCs into chronic ischemic rats (4 weeks after ischemia), a significant portion (approximately 57%) of the CFDA-SE+ cells that were found in the brain were CD25+ cells (Fig. 9A, B). In contrast, we could not detect any CFDA-SE+ cells coexpressing CD29, indicating that few MSCs in the injected BMC population infiltrated into the chronic ischemic brain (Fig. 9).
Quantitation of CD25+ cells that infiltrated the chronic ischemic brain. The MCAO rats were injected with CFDA-SE-labeled MSCs both IV and IA at 4 weeks after MCAO induction and sacrificed 3 h posttransplantation for analysis. (A) Many infiltrated BMCs (CFDA-SE+, green) were colabeled with CD25+ antibody (red). Scale bar: 10 μm. (B) The percentage of CD25+ cells among the total BMCs that infiltrated the brain was calculated by dividing the number of CFDA-SE/CD25-double positive cells by the number of total CFDA-SE+ cells that were found in the brain. N = 5. The percentage of CD29+ cells among the infiltrated BMCs (CFDA-SE+ cells) was also indicated at the bottom of the table.
The high percentage of CD25+ cells among the total infiltrating BMCs indicates that these CD25+ cells (which include regulatory T-cells) may confer the beneficial effects detected after BMC transplantation. Perhaps these cells at least partially function by suppressing the activation of microglial cells that exist in the chronic ischemic brain.
Discussion
Although the beneficial effects of BMC transplantation have been studied during the acute stage of stroke (6,9, 12,18,23), it is unclear whether transplanted BMCs exert any beneficial effects on chronic ischemic stroke. To examine the effects of BMC transplantation in a rat model of chronic stroke, we first delivered BMCs at 5 weeks post-stroke via both IA and IV routes. Additionally, BMCs were also transplanted (via IV injection) at 3 consecutive weeks, 10, 11, and 12 weeks, after ischemia, to maximize any potential efficacy of BMC transplantation.
Our results demonstrated that the BMC transplantation improved behavioral performance in both mNSS and foot fault tests. Intriguingly, significant differences between the BMC-treated and control groups were observed from 7 weeks in mNSS and 1 week (the first measurement) posttransplantation in the mNSS and foot fault test, respectively. Currently, it is not clear what causes the immediate positive effect in the foot fault test. We speculate that this initial behavioral improvement may be due to revival of dying neurons or activation of neural activity by cytokines released from the transplanted BMCs. However, the immediate behavioral improvement seems to be insignificant so that only sensitive behavioral tests, such as the foot fault test, but not mNSS, could detect it.
In addition to the functional recovery, we also detected increased parenchymal brain volume by the BMC transplantation. Taken together, the functional and structural recovery we observed indicated that BMC transplantation was effective during the chronic stage of ischemic stroke.
Usually, the beneficial effect of the transplanted BMCs during the acute stage of stroke could be mediated by the protection of dying brain cells and the promotion of neural proliferation. Our experiments were performed during the chronic stage of ischemia, in which a large portion of the brain cells in the ischemic region have been already damaged, so we speculated that a large portion of the functional and structural recovery that was observed in the BMC-treated group was caused by BMC-mediated neural proliferation.
Although ischemia-induced neural proliferation has been observed during the acute stage of ischemic stroke (3,20), no information describing the effects of BMC transplantation on neural proliferation during the chronic stage of stroke is available.
In this study, we demonstrated that active neural proliferation was evident in the BMC-treated chronic ischemic rats in both the SVZ and the dentate gyrus, which are the two major neurogenic regions in the brain. This increase in neural proliferation was not likely caused by the ischemic injury itself because ischemia-induced neural proliferation lasted only until 2 weeks poststroke (4,33). In addition, we could not detect such a massive increase in neural proliferation when vehicle (0.1 M PBS) was injected into the chronic ischemic rats. These results suggest that BMC transplantation enhanced neural proliferation in the chronic ischemic brain, and it may have at least partially contributed to the recovery in the brain during the chronic phase of ischemic stroke.
Based on previous studies showing that inflammation confers negative effects toward neural proliferation in the injured brain (10,16,22,29), we speculated that the increased neural proliferation due to BMC transplantation in the chronic ischemic brain may be linked to the suppression of inflammation by BMCs. In support of this, BMC administration drastically reduced the number of activated microglia in the peri-infarct striatum and dentate gyrus of the chronic ischemic brain (Fig. 7). This result suggests that inflammation reactions still occur even during the chronic stage of stroke, and BMC transplantation efficiently reduced inflammation leading to enhanced neural proliferation. In line with this interpretation, we and other researchers showed that administration of minocycline after ischemic brain injury drastically reduced the number of activated microglia and increased neural cell proliferation in the chronic ischemic brain (22) (Fig. 8). Intriguingly, however, a recent study using a focal traumatic brain injury model showed no clear correlation between minocycline-mediated decrease of activated microglia and enhancement of neurogenesis (25), which indicates that the effects of anti-inflammation on neural proliferation are rather complicated and are possibly affected by a variety of factors, such as the nature, location, or extent of brain injury, experimental paradigms, and the degree of microglial activation.
The mechanism underlying the anti-inflammatory role of the transplanted BMCs is unclear. Previous studies have suggested that BMCs can reduce microglial activation by secreting anti-inflammatory cytokines (28). Furthermore, regulatory T-cells that exist within the BMC population have been shown to exert a major role in antiinflammatory processes during the acute ischemic stage by reducing activated microglia (21). Therefore, it is tempting to speculate that CD25+ regulatory T-cells may play a crucial role in the anti-inflammatory processes that occur in the chronic stage of stroke. Consistent with this speculation, we detected that approximately 57% of the infiltrating BMC-originated cells in the chronic ischemic brain were CD25+ cells. This result was surprising because the percentage of CD25+ cells that were present in the BMC population that was used for transplantation was very low (approximately 0.36%) (Fig. 1B). This suggests a potentially significant role of CD25+ cells (which include regulatory T-cells) in the recovery from ischemic injury during the chronic stage of stroke.
We also examined whether bone marrow-derived mesenchymal stem cells infiltrated into the chronic ischemic brain and contributed to the beneficial effects observed by BMC transplantation. As shown in Figure 9B, we failed to detect any CD29+ (a marker for leukocytes, granulocytes, and mesenchymal stem cells) cells among CFDA-SE+ cells found in the ischemic brain. This result indicated that bone marrow-derived mesenchymal stem cells in the BMC population were not responsible for the recovery observed after BMC transplantation. In addition, we could not detect CFDA-SE+ cells expressing neuronal markers at 7 days after transplantation, indicating no neuronal differentiation of the transplanted BMCs (data not shown).
In contrast to our results, a previous study reported that the therapeutic window for BMC transplantation was only 72 h after the onset of stroke, and BMC injection 1 week poststroke did not show any difference in behavioral performance (34). Explanations for this discrepancy may include either the difference in method of BMC delivery [e.g., Yang et al. (34) used IV injection, and our experiment used both IV and IA injections] or the time window for behavioral analysis [e.g., Yang et al. (34) assessed animals only up to 4 weeks posttransplantation, and our experiment assessed up to 8 weeks posttransplantation]. In fact, we detected BMC-mediated behavioral improvements in the mNSS test from 7 weeks after the BMC transplantation, which suggests that long-term follow-up analysis may be needed to detect a significant beneficial effect when BMCs are administered to chronic stroke rats.
At this stage, it is not clear if the additional BMC administrations at weeks 10, 11, and 12 post-MCAO surgery played a critical role in recovery from chronic ischemic brain injury. The first BMC transplantation (at 5 weeks poststroke) seemed to be sufficient in enhancing neural proliferation because new neural cells were labeled with BrdU (7~9 weeks, daily) a week before the second BMC transplantation (at 10 weeks) (Figs. 3A, 5B and C, 6B and C).
However, it could be possible that additional BMC injections contributed to the prominent beneficial outcomes of BMC transplantation possibly by providing a better environment for the survival or maturation of the new neural cells (i.e., by offering growth factors or cytokines and reducing inflammation in the injured brain). Therefore, it would be of interest to investigate whether or by what mechanism(s) the additional IV administrations of BMCs played a role in recovering from the chronic ischemic brain injury.
Intriguingly, the BMC fraction composition was changed by ischemic induction and at different time points after the ischemic injury (Fig. 1). This finding suggests that the therapeutic effects of BMC transplantation may vary depending on the source of the cells (i.e., whether the cells are derived from a healthy donor or a stroke patient and also when the cells are obtained after a stroke). Consistent with our observation, a recent study demonstrated that BMSCs that were derived from ischemic rats displayed better therapeutic effects than those that were derived from normal rats in the acute stage after MCAO (35). The detailed mechanisms by which the composition of the BMC population affects the therapeutic efficacy, especially at various stages of stroke, awaits further investigation.
Although transplanted BMCs have been tracked at the acute stage of stroke (6), no information is available regarding the distribution of transplanted BMCs during the chronic stage of stroke. In our tracking experiment, we observed that the highest number of labeled BMCs infiltrated the chronic ischemic brain 3 h after BMC administration. Interestingly, the number of infiltrating BMCs decreased dramatically between 3 h and 1 day posttransplantation and remained at a comparable level thereafter. This pattern of a rapid decrease in infiltrating BMCs was also observed in the acute stage of stroke (6). Although the cause of the rapid disappearance is unclear, a previous study of acute stroke demonstrated that many infiltrating BMCs were terminal deoxynucleotidyl transferase dUTP nick-end labeling positive (TUNEL+) at 3 h postinjection (6). Therefore, it is plausible that most of the infiltrating BMCs died within a day after transplantation, and only a small population of remaining BMCs contributed to the recovery that was observed in the chronic stroke rats in our experiment.
Taken together, our study showed that BMC transplantation during the chronic stage of stroke conferred beneficial effects on both brain structure and behavioral performance. The prominent changes observed in the BMC-transplanted chronic ischemic rats were reduced inflammation and enhanced neural proliferation in the brain. Strikingly, we determined that a large portion of the infiltrating (CFDA-SE labeled) BMCs in the brain were CD25+ cells because approximately 57% of the CFDA-SE+ cells in the brain were colabeled with CD25 signal (Fig. 9A, B). The percentage of CD25+ cells in the original BMC population used for injection was only about 0.35%; this suggests that CD25+ cells seem to infiltrate into the chronic ischemic brain very efficiently compared to the other cell types in the BMC population. Based on these results, we propose a working model in which infiltrating CD25+ cells suppress inflammatory responses (i.e., microglial inactivation) in the chronic ischemic brain, which thereby leads to enhanced neural proliferation and structural and functional recovery.
Overall, our study provides valuable insight into a BMC-mediated therapeutic approach to treat patients with chronic stroke. These findings regarding the mechanisms underlying the function of BMCs should lead to the development of an effective therapeutic regimen to treat chronic stroke patients who do not currently have any satisfactory therapeutic options.
Footnotes
Acknowledgments
This research was supported by the Stem Cell Research Program (2010–0020407) and the Bio and Medical Technology Development Program (2012M3A9C7050130) of the National Research Foundation funded by the Ministry of Science, ICT and Future Planning and a research grant (A120254–1201–0000200) from the Ministry of Health & Welfare, Republic of Korea. The authors declare no conflicts of interest.