
Abstract
Clinical studies have proved the therapeutic potential of hepatocyte transplantation as a promising alternative to whole organ liver transplantation in the treatment of hereditary or end-stage liver disease. However, donor shortage seriously restricts cell availability, and the lack of appropriate cell culture protocols for the storage and maintenance of donor cells constitutes a significant obstacle. The aim of this study was to stimulate mature hepatocytes in culture to multiply in vitro and track their fate on transplantation. Rat hepatocytes isolated nonenzymatically were cultured serum free for up to 10 days. They were stimulated into proliferation in the presence of growth factors and conditioned media from nonparenchymal and hepatocyte culture supernatants, as well as 10 mM lithium chloride (LiCl). Cell proliferation was assessed by determining DNA content. Additionally, the extent of cell differentiation was estimated using immunofluorescence staining of hepatic, biliary, progenitor, and mesenchymal markers and gene expression analyses. Transplantation studies were performed on the Fischer CD26-mutant rat following pretreatment with retrorsine and partial hepatectomy. Proliferating hepatocytes increasingly adopted precursor characteristics, expressing progenitor (OV6, CD133), hepatic lineage (CK18), biliary (CD49f, CK7, CK19), and mesenchymal (vimentin) markers. The supplement of LiCl further enhanced the proliferative capacity by 30%. Transplantation studies revealed extensive repopulation by large donor hepatocyte clusters. Furthermore, bile duct-like structures deriving from donor cells proved to be immunoreactive to ductular markers and formed in close proximity to endogenous bile ducts. Mature hepatocytes reveal their potential to “switch” between phenotypes, adopting progenitor characteristics during proliferation in vitro. Following transplantation, these “retrodifferentiated” cells further expanded in vivo, thereby generating bipotentially differentiated progenies (hepatocytes and bile duct-like structures). This apparent plasticity of mature hepatocytes may open new approaches for cell-based strategies to treat liver disease.
Keywords
Introduction
The unique ability of the liver to regenerate has been common knowledge since as early as the eighth century BC, even featuring in Greek mythology. Recent translational research in cell transplantation has demonstrated liver regeneration by proliferating and implanting mature hepatocytes into the host liver of animal models. Moreover, clinical studies have demonstrated the therapeutic potential of transplanted mature hepatocytes in the treatment of hereditary metabolic liver disease (27) and acute liver failure (2). However, the limited availability of suitable cell sources and the lack of standardized protocols for cryopreservation have hindered the procedure in its progression from the laboratory to the clinic. Furthermore, the unabated mitotic ability that hepatocytes display in vivo is rapidly lost under most cell culture conditions.
While the majority of studies have concentrated on the differentiation of hepatocytes during proliferation in culture, numerous strategies have attempted to supply cells for transplantation from different sources to overcome the burden caused by limited numbers of donor organs. These studies have focused, for instance, on fetal liver cells and mesenchymal stem cells for therapeutic purposes (24,37).
Mitaka et al. were the first to succeed in culturing hepatocytes under serum-free conditions, whereby mature hepatocytes briefly underwent clonal expansion (31). Furthermore, Tateno et al. demonstrated that the interaction between hepatocytes and nonparenchymal cells in vitro has a protective role in cell differentiation, since cells grow for longer periods in this setting (50). Besides, hepatocytes in the vicinity of nonparenchymal cells are able to express biliary markers such as cytokeratin 7 (CK7) and CK19 (29). Most recently, lineage-tracing studies demonstrated that bipotent liver progenitor cells may arise from mature hepatocytes in culture (6).
We previously demonstrated that serum-free-cultured mature hepatocytes are able to repopulate the host liver after transplantation, despite having lost some hepatocyte-specific characteristics in vitro (1). More recently, we established a cell culture system in which primary hepatocytes are stimulated into rapid proliferation in the presence of growth factors and conditioned media from nonparenchymal cell and hepatocyte culture supernatants (17). In the present study, we further improved this culture protocol by adding lithium chloride (LiCl), resulting in enhanced expression of biliary, mesenchymal, and progenitor characteristics.
The aim of the study was to investigate the fate of these generated “retrodifferentiated” hepatocytes following transplantation in vivo. Employing the established syngeneic cluster of differentiation 26 (CD26)-deficient Fischer 344 (F344) rat model, CD26-positive cultured cells were transplanted into the livers of CD26-negative recipient animals, enabling the detection of transplanted cells using immunofluorescence staining techniques. Cell division in endogenous hepatocytes was blocked by retrorsine, a pyrrolizidine alkaloid, to provide transplanted cells with a growth advantage. As a result, these retrodifferentiated cells did not only regain their mature hepatocyte attributes when integrating and further expanding in the host liver, but also generated bile duct-like structures.
Materials and Methods
Animals and Reagents
Male CD26-positive F344 donor rats were purchased from Charles River (Sulzfeld, Germany). Recipient male F344 rats (deficient in CD26) were bred from parent animals kindly provided by E. Laconi from the Istituto di Patologia Sperimentale, Cagliari, Italy. Animals were kept on a 12-h day/night rhythm and had a standard diet (ssniff, Soest, Germany). All animal breeding, care, and experimentation procedures were performed in accordance with German national and regional legislation on animal protection and institutional approval.
Epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), and calf serum (NCS) were supplied by Biochrom (Berlin, Germany). Williams E medium (5 mM glucose) and holotransferrin were purchased from Applichem (Darmstadt, Germany). Bovine insulin was obtained from Serva (Heidelberg, Germany) and Percoll from Pharmacia (Freiburg, Germany). Culture dishes were acquired from Sarstedt (Wiesbaden, Germany). Unless specified otherwise, all other chemicals were supplied by Sigma-Aldrich (Deisenhofen, Germany).
Primary antibodies were purchased and used as depicted in Table 1. Secondary fluorescence labeling antibodies (Alexa Fluor) were obtained from Molecular Probes (Goettingen, Germany).
Antibodies for Colocalization Studies

CD26, cluster of differentiation 26; CK7, cytokeratin 7; OV6, oval cell marker 6. BD Pharmingen (Heidelberg, Germany); Sigma-Aldrich (Deisenhofen, Germany); Dako (Hamburg, Germany); Pro Gen (Heidelberg, Germany); Serotec (Puchheim, Germany); R&D Systems (Wiesbaden-Nordenstadt, Germany); Santa Cruz (Heidelberg, Germany).
Isolation and Culture of Hepatocytes
Hepatocytes were isolated from CD26-positive, male F344 rats (200–250 g body mass) using a nonenzymatic isolation protocol to avoid contamination by nonparenchymal liver cells, according to Meredith (26). Subsequently, the liver was perfused with 2,000 ml of calcium-free Krebs–Ringer buffer for 40 to 50 min without recirculation and then minced with forceps in the buffer. Cells passed through an 80-μm nylon filter (BD Falcon, Bedford, MA, USA), and hepatocytes were separated in a centrifuge at 10 × g for 3 min. Hepatocytes were freed of debris and dead cells by Percoll (GE Healthcare, Uppsala, Sweden) centrifugation and washed once again with Williams E medium.
Proliferation studies were carried out using 5,000 or 14,000 cells/cm2 plated on 35-mm dishes. Isolated cells were plated onto collagen gel matrices (17,40) in Williams E medium with 5 mM glucose, supplemented with 4% NCS, 0.1 mM dexamethasone, and 1 nM insulin. After 3 h, the cells were washed, and the medium was changed to one of three different combinations of serum-free media, which were subsequently changed daily, 1.5 ml/ dish. Basal medium (BM) consisted of Williams E medium supplemented with 0.2% bovine serum albumin (BSA), 15 mM HEPES, 0.2 mM ethanolamine, 0.2 mM phosphoethanolamine, 5 μg/ml transferrin, 10 μg/ml heparin, 0.1 μM hydrocortisone, 0.1 μM dexamethasone, 0.2 μM insulin, 75 nM sodium selenite, 5 ng/ml bFGF, 10 ng/ml EGF, 17 mM penicillin, and 16 mM streptomycin. The complete medium (CM) comprised a mixture of 50% BM enriched with 20% hepatocyte-conditioned medium (HCM) and 30% stromal cell-conditioned medium (SC-CM). The preparation of conditioned media (HCM and SC-CM) from hepatocyte culture (collection after 24 h) and non-parenchymal cell supernatants (collection after 48 h) was described in detail before (17) as well as the preparation of the collagen gel matrices based on the solubilization of rat tail collagen and its cross-linking with glutaraldehyde (17,40). Further growth stimulation was achieved through the addition of 10 mM LiCl (Applichem, Darmstadt, Germany) to CM (20).
In preparation of the transplantation experiments, 14,000 cells/cm2 were seeded in large culture dishes 60 mm in diameter and stimulated to proliferate in the three different media (BM, CM, and CM + LiCl). The conditions provided by CM + LiCl proved to yield the greatest number of cells as well as the most marked change to the desired progenitor profile based on marker expression. Thus, only these cells were selected for the in vivo studies.
Detachment of Cultured Hepatocytes
Cells were cultivated for 8 days on 60-mm dishes, then washed three times, and incubated for 15 min at 37°C in 2.5 ml HBSS (without Ca2+ Mg2+), supplemented with 0.02 mM EDTA. Subsequently, cells were washed once with HBSS (with Ca2+ Mg2+) supplemented with 0.02% BSA and next incubated in 3 ml HBSS + BSA with collagenase (3 mg/ml) for 5 min at 37°C. Each individual dish was checked microscopically for cell detachment.
Incubation was stopped immediately with 5 ml RPMI-1640 supplemented with 10% FCS. Detached cells were collected and diluted with RPMI (50-ml Falcon tube; BD Falcon) supplemented with 10% FCS. The gels with remaining cells were minced and resuspended by mixing in RPMI + 10% FCS. Thereafter, cells were filtered through a 70-μm cell strainer (BD Falcon) into a second 50-ml tube. Cells were then pelleted for 5 min at 50 × g at 4°C and gently resuspended in 4 ml Cell Dissociation Solution Nonenzymatic 1×. After incubation for 6 min at 37°C, cells were resuspended in 20 ml RPMI + 10% FCS, filtered through a 70-μm strainer, diluted to 50 ml with RPMI + 10% FCS. Afterwards, cells were centrifuged for 4 min at 30 × g and 1 min at 60 × g and finally resuspended in 1 ml pure RPMI. Detached cells from one dish yielded approximately 1 × 106 cells and were transplanted when displaying vitality greater than 85% (trypan blue exclusion).
Transplantation
Male recipients (9–12 weeks) were injected with retrorsine (30 mg/kg; Sigma-Aldrich, Germany) 6 and 4 weeks prior to transplantation. Animals were anesthetized under constant sevofluran (Sevorane®, Abbott, Wiesbaden, Germany) inhalation.
Carprofen (Rimadyl®, Pfitzer, Berlin, Germany) was applied subcutaneously during anesthesia (30 mg/ kg body weight). During the 30% hepatectomy, the lateral segment of the left liver lobe was removed by central ligation. Cells of six dishes (diameter 60 mm) were injected slowly over 2 min into the portal vein using a 28-gauge syringe. Recipient rats were euthanized 24 h and 16 weeks following transplantation, respectively.
Estimation of Cell Proliferation
Cell proliferation was estimated by determining the incorporation of methyl-[3H]thymidine as well as DNA content and quantified using established protocols (40).
Immunofluorescence Staining Analysis
Cytospins were obtained from freshly isolated hepatocytes from donor rats and fixed in ice-cold acetone for 10 min. Slides were stored at 8°C until staining. Cultured cells were fixed after 8 days directly on the dishes. Cells were washed three times with PBS within the culture dishes, fixed in acetone, and stored at 4°C. Tissue samples from recipient livers were excised and snap frozen in 2-methylbutane at −70°C. Cryostat sections (5 μm) were fixed in ice-cold acetone for 10 min and stored at −80°C.
For immunofluorescence staining, the primary antibodies anti-connexin 32 (Cx32), -CK18, -CD49f, -CK7, -oval cell marker 6 (OV6), or -vimentin (Table 1) were incubated overnight at 4°C. In preparation for the immunofluorescence colocalization investigations, the primary antibody CD26 was combined with either anti-Cx32, -CK18, -CD49f, -CK7, -OV6, or -vimentin antibodies and incubated overnight at 4°C (Table 1). The secondary antibodies Alexa 488-conjugated goat anti-rabbit IgG and Alexa Fluor 555 goat anti-mouse IgG (1: 400, 1 h at RT) were used for immune labeling. Samples were covered with Vectashield® mounting medium plus 4′,6-diamidino-2-phenylindole (DAPI; 4 μl/ml; Vector Laboratories, UK) and evaluated under a microscope (LEICA DM IRE2, Bensheim, Germany).
Relative Quantification Using RT-PCR Analysis
Total RNA was isolated with the RNeasy mini kit (Qiagen, Hilden, Germany) and stored at −80°C. First-strand cDNA was synthesized using an iScript cDNA Synthesis Kit (BioRad, Munich, Germany) with oligo-(dT)12–18. PCRs (iQ Sybr Green Supermix, BioRad, Munich) were carried out using an iCycler (BioRad, Munich) with intron spanning primers (Eurofins MWG, Ebersberg, Germany), (Table 2) and reactions were normalized to β2-microglobulin (β2-MG) and hypoxanthineguanine phosphoribosyltransferase (HPRT) as internal standards. Amplification and expression analysis were performed as previously described (20).
Primers for Expression Studies

VIM, vimentin; HPRT, hypoxanthine-guanine phosphoribosyltransferase; β2MG, β2-microglobulin.
Results
Proliferation and changes in phenotype of adult rat hepatocytes in vitro have been previously reported in Wistar strain rats (17). Since our syngeneic transplantation experiments required the use of Fischer rats, we adjusted and optimized the settings of cell culture to the F344 rat strain and tested the proliferative potency and reexpression of progenitor markers in cultured F344 hepatocytes. The aim of the study was to develop an optimal cell culture system producing progenitor-like cells to demonstrate proof of principle of bipotent differentiation in the retrorsine-based rat model for liver repopulation.
Lithium is known to influence the wingless-related mouse mammary tumor virus integration site (wnt) pathway while inhibiting glycogen synthase kinase 3β (GSK3β) (9,16). The inhibition of GSK3β induces the wnt pathway, which is crucial for proliferation and growth of embryonic cells as well as cell proliferation in adult tissues. Lithium also enhances the proliferation and differentiation of neural progenitor and stem cells while protecting neural tissue from hypoxic–ischemic injury (23). We also chose LiCl to enhance proliferation in our cell culture system to maximize the yield in cell number for downstream transplantation studies.
The Influence of Culture Conditions, Plating Density, and Lithium Chloride on Cell Growth in Culture
Hepatocytes were seeded at low [5,000 cells/cm2, as in Wistar cultures (17)] and higher (14,000 cells/cm2) densities. Using the smaller 35-mm dishes for proliferation studies, the higher seeding density only led to moderate growth in BM (Fig. 1A). The use of CM, which contained additional mitogenic factors from conditioned media, led to extensive and rapid proliferation from day 2 to day 7. As previously demonstrated (20), LiCl exerted a powerful proliferative effect on cultured hepatocytes, and in the presence of 10 mM LiCl (added daily), proliferation was further increased from day 5 to day 10. The final DNA content (parameter for cell growth) on day 10 was augmented by approximately 30% when compared with control cultures without LiCl. Cell growth kinetics did not differ between 35-mm and 60-mm dishes (Fig. 1B). We therefore used a higher seeding density to increase the cell yield for transplantation and employed larger 60-mm dishes for faster and easier handling.
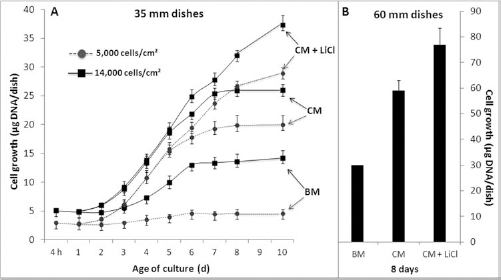
Cell growth of F344 hepatocytes in culture. (A) Time course of hepatocyte growth on 35-mm dishes with a low or high seeding density as indicated in different media: Basal medium (BM) based on Williams E medium supplemented with bovine serum albumin (BSA), epidermal growth factor (EGF), and basic fibroblast growth factor (bFGF), the complete medium (CM) comprised a mixture of 50% BM enriched with 20% hepatocyte-conditioned medium (HCM) and 30% stromal cell-conditioned medium (SC-CM); in further experiments, CM was also supplemented with lithium chloride (LiCl) to enhance proliferation. (B) Cells cultured on 60-mm dishes at a seeding density of 14,000 cells/cm2. Data are mean ± SD of three different cell preparations. In each single experiment, all culture conditions were examined in parallel using hepatocytes from the same batch.
Changes in the Phenotype of Hepatocytes Proliferating In Vitro
Cultured cells were stained immunochemically with hepatic, progenitor, and mesenchymal markers to investigate cytoskeleton structures as well as other changes in differentiation features.
As demonstrated previously employing hepatocytes from Wistar rats (17), freshly isolated hepatocytes from F344 rats stained positive for the hepatocellular differentiation marker CK18 as well as for the functional marker Cx32 (a component of hepatic gap junctions). However, freshly isolated hepatocytes did not express the bile duct marker CK7, the biliary progenitor marker CD49f (integrin α6), the mesenchymal marker vimentin, or the progenitor marker OV6.
After 8 days of culture, the hepatocellular marker CK18 (Fig. 2A) was distributed in the cytoplasm with areas of intense staining in parts of the cultured cells. Visible mitotic figures (nuclei) indicated regular cell division (Fig. 2A, insert). The gap junction protein Cx32 could still be visualized, but it was not distributed in any normal manner within the cell membranes (Fig. 2B); the staining appeared as tiny spots around and within the cell nuclei. Additionally, the cells were now positive for CD49f (Fig. 2C), CK7 (Fig. 2D), the oval cell marker OV6 (Fig. 2E), and vimentin (Fig. 2F). As expected, the markers CD49f and OV6 were expressed in the plasma membrane, whereas CK7 and vimentin as cytoskeletal markers were stained in the cytoplasm. CD49f was distributed rather uniformly in contrast to the CK7 staining, which appeared in cell clusters. Vimentin demonstrated areas with more or less intensive staining heterogeneously distributed throughout the cell plates.

Immunofluorescence staining of differentiation markers in cells cultured for 8 days in CM with LiCl. Cells were fixed and processed on the dish. Nuclear counterstaining with 4′,6-diamidino-2-phenylindole (DAPI) in blue. (A) Cells were stained for cytokeratin 18 (CK18) determining the hepatocytic cell linage. Inset: Single cells show different stages of mitosis. (B) The atypical pattern of connexin 32 (Cx32) distribution. Cx32 is dotted around the nuclei but was not membrane bound. (C–F) Immunofluorescence staining as marked. Original magnifications as indicated by the scale bar: 50 μm. CD49f, cluster of differentiation 49f; OV6, oval cell marker 6.
QRT-PCR Analysis
Gene expression analyses for the differentiation markers CK18, Cx32, CK19, CK7, vimentin, and CD133 were assessed in a time-dependent manner in cultured cells up to 8 days (Fig. 3). Freshly isolated hepatocytes exhibited only low concentrations of CK18 mRNA compared to cells in culture. Throughout the duration of culture, CK18 mRNA levels were devoid of major variations, whereas LiCl enhanced the mRNA level from day 1. After 24 h in culture, the expression of Cx32 decreased to a third of its original value compared to fresh isolated hepatocytes; from the third day onward, it continually decreased to less than 10% of the initial value. LiCl did not play a significant role in this process. The bile duct marker CK19 was almost not detectable in freshly isolated cells; its expression increased slightly from day 4 to day 8. The supplementation of LiCl enhanced this effect up to sixfold until the endpoint of the experiment. The expression of the biliary marker CK7 was absent in freshly isolated cells. However, from day 1 to day 8 in culture, the initial value increased by a factor of 2 × 104. LiCl had no influence on the expression of CK7. The mesenchymal marker vimentin was only marginally expressed in freshly isolated cells. Afterwards, the mRNA level increased up to 2,400-fold on day 8 and was even further enhanced by LiCl. The stem cell marker CD133 was almost not expressed in freshly isolated cells, but could be detected as early as on the first day of culture. After an initial 2 days in culture, expression increased due to addition of LiCl; however, it reduced afterwards and finally stagnated.

Relative RT-PCR quantification of gene expression profiles of proliferating hepatocytes at different points in time of culture. Cultured cells were incubated in CM without LiCl (gray columns) or in CM supplemented with 10 mM LiCl (black columns). Analysis of hepatocytic (CK18, Cx32), bile duct (CK19, CK7), mesenchymal (vimentin), and stem cell (CD133) markers on the mRNA level. Freshly isolated hepatocytes set at time 0. Data are mean ± SD representatives of three different investigations.
To summarize, hepatocytes retained their hepatocytic lineage marker CK18 during the entire course of cell culture. However, the expression of the differentiation marker Cx32 decreased from day 3 onward, the point in time at which cells started to proliferate. On day 4 of culture, by which cells reached their exponential growth rate, expression levels of biliary markers such as CK19 and CK7 and of the mesenchymal marker vimentin massively increased. The expression of the stem cell marker CD133 was not affected by the proliferation rate, but was considerably enhanced for the two first days of culture by the addition of LiCl.
Bipotent Liver Repopulation Following Transplantation of Cultured Cells
Cells were detached from the culture plate following 8 days in culture and then transplanted into the rat liver repopulation model (Fig. 4). Sixteen weeks after transplantation, clusters of hepatocytes displaying donorspecific CD26 appeared in close proximity to the portal fields and had greatly increased in size, now expanding throughout the liver lobes (Fig. 4A). On average, these clusters consisted of approximately 50 to several hundred cells in diameter grossly displaying hepatocytic features with seamless integration into the recipient liver parenchyma. The staining pattern of CD26 displayed its typical distribution in the basolateral bile canalicular domains, which points to completely polarized, differentiated, and metabolically active hepatocytes (Fig. 4D). Cx32 was distributed as points in the cell membrane of adjacent donor cells as well as on adjacent endogenous hepatocytes. This typical staining pattern of gap junctions represented the normal morphology of fully integrated hepatocytes. In contrast, some donor cells appeared to behave differently, since their expression of the specific CD26 adopted a more compact (cytoplasmic) pattern and they lacked the coexpression of Cx32 (Fig. 4A, inset). In particular, these donor-derived cells appeared as bile duct-like structures and were regularly seen in one of six to eight view fields.

Representative overviews of host liver repopulation by transplanted cells cultured for 8 days in CM with LiCl. Multilayer immunofluorescence micrographs with CD26 identified in red, gap junction protein Cx32, or biliary progenitor marker CD49f in green, and cell nuclei are visible with DAPI in blue. (A) Repopulation of a liver section as seen 16 weeks after transplantation. Large and in part confluent clusters of cells derived from cultured cells are detectable; the image contains seven merged adjacent microscopic fields. Representative area demonstrating donor cell clusters counting 100 to 300 cells in diameter. Scale bar: 100 μm. The inset depicts a duct-like structure emerging from transplanted cells. (B) Expression of donor-specific and membrane-bound CD26 on freshly detached hepatocytes with minor and irregular Cx32 staining. (C) Transplanted cells close by the portal vein (PV) displaying donor-derived CD26. (D) Donor cell cluster emerging from the portal fields. The cells are well dotted with Cx32, indicating perfect integrated hepatocytes. (E) CD26 staining combined with membrane-bound CD49f on detached cultured cells prior to transplantation. (F) CD49f/CD26 double-stained small groups of transplanted cells close to the PV shortly after transplantation. (G) Hepatocytes derived from cultured cells revealing a pattern of star- and garland-like structures of CD26 staining. CD49f/CD26 double-stained ductlike structures derived from cultured cells in close proximity to the PV. Original magnifications as indicated by the scale bars: 50 μm. (H) At 24 h following transplantation, CD26-positive cultured cells still coexpressed the mesenchymal marker vimentin while entering the hepatic parenchyma. Original magnification as indicated by the scale bar: 25 μm.
To investigate the quality and extent of liver cell repopulation in more detail, we assessed the changes in marker expression starting from the cells detached after 8 days in culture suspension and following transplantation in vivo. Freshly detached cells homogenously displayed the donor-classifying hepatic marker CD26 but did not stain for Cx32 (Fig. 4B). Furthermore, cells stained intensively positive for CD49f as progenitor and bile duct marker (Fig. 4E). Twenty-four hours following transplantation, donor cells had traversed the endothelial barrier and just entered the parenchyma. At this point in time, they were still negative for Cx32 (Fig. 4C); however, they expressed CD49f diffusely (Fig. 4F). Sixteen weeks later, most donor-derived cells had formed large clusters. However, some CD49f-positive donor cells were sporadically disseminated in between these clusters and had arranged into ductular structures (Fig. 4G). They were found predominantly in close proximity to the endogenous bile duct system, emerging from the portal areas. Immunofluorescence colocalization revealed staining of vimentin in donor cells only at the early time point (24 h) posttransplantation (Fig. 4H). At this point in time, the cells had not yet completely integrated in the recipient liver but had already left the hepatic sinusoids. Sixteen weeks later, the donor-derived bile duct-like cells were negative for vimentin and had therefore lost the criterion for the progenitor phenotype (data not shown).
Formation of Duct-Like Structures
The expression pattern of progenitor/bile duct markers in donor-derived cells was elucidated by immunofluorescence colocalization studies (Fig. 5). Sixteen weeks after the transplantation of cultured cells, donor-derived cells formed single (Fig. 5C, L) or multiple ducts representing a tubular system (Fig. 5F, I). Within the host liver parenchyma, these CD26-positive cells did not exhibit Cx32 (as seen Fig. 4A, insert) but colocalized with CD49f (Fig. 5A–C and D–F) and CK7 (Fig. 5G–I) as well as with OV6 (Fig. 5J–L). CD26 staining was detected inside the lumen, whereas the markers CK7, CD49f, and OV6 were stained outside the lumen and around the ducts. Taking into account the localization, morphology, and distinct marker expression, the ductular structures derived from donor cells may resemble newly formed bile ducts within the recipient liver. As there is an overlap of progenitor and bile duct markers, the state of differentiation cannot be determined beyond doubt. However, the arrangement in ductular systems and the lack of vimentin expression suggests the more mature bile duct feature.
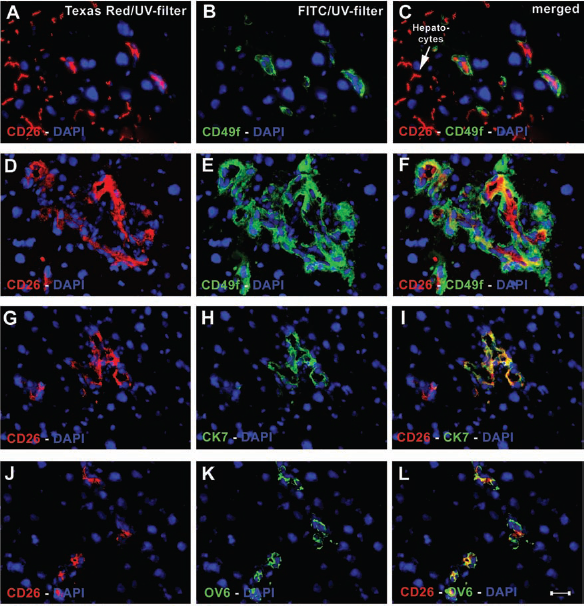
Colocalization of differentiation markers in duct-like structures by multilayer immunofluorescence staining 16 weeks after transplantation. Fluorescein isothiocyanate (FITC) and tetramethylrhodamine isothiocyanate (TRITC) fluorescence channels are presented separately and merged images on the right column. (A–C) Donor cell coexpressing the biliary progenitor marker CD49f in close proximity to integrated donor-derived hepatocytes in the host liver. CD26 staining indicates the host liver origin (descendants from cultured cells). (D–F) Duct-like structures stained inside the lumen with CD26 and surrounded by staining of CD49f. (G–I) The biliary marker CK7 was detected on duct-like structures coexpressing CD26. (J–L) The progenitor marker OV6 stains positive outside around the ducts. Yellow staining indicates overlay. Scale bar: 50 μm.
Discussion
Hepatocytes Proliferating In Vitro Share Common Features with Hepatic Progenitor Cells
The role of nonparenchymal cells (31,32,50), cell– matrix interactions (10,15), humoral impacts (14), cell membrane factors (34), and the interaction between cell types in culture (30) on the differentiation and proliferation of mature hepatocytes has been thoroughly investigated. Biliary markers (e.g., CK19, CD49f) also overlapping with hepatic progenitor markers can be detected in isolated hepatocytes (mostly transcript levels) and for quite some time in primary cultures (protein level or even enzyme activity) (3,7,46). The cell isolation techniques yielded purity, and culture conditions for these cells grossly vary, and several studies actually reported transdifferentiation of adult hepatocytes into biliary cells in vitro (29,35,36). In accordance with previous data, our experiments confirm clear and ever-increasing expression of bile duct and progenitor markers CK7 and CK19 during the course of culture (17). In our experimental settings, mature hepatocytes were stimulated into proliferation employing collagen gel-coated dishes and medium supplemented with hepatocyte-specific mitogens obtained from conditioned media of hepatocytes as well as stromal cells. This effect is in line with observations that small hepatocytes stain positively for the markers CK7 and CK19 when attached to nonparenchymal cells in culture (33,50).
During the course of cell culture, hepatocytes appeared to switch their phenotype, gradually adopting a bipotent progenitor cell feature. This was characterized by retention of the hepatic lineage marker CK18 and expression of various bile ducts as well as some classic progenitor markers. Hepatoblasts from fetal liver development and the intrahepatic stem cells “oval cells” are also known to be bipotent. The latter cell type represents the most extensively investigated population of progenitor/stem cells in the adult liver. They are released during advanced chronic liver injury, whenever the replication ability of adult hepatocytes is massively impaired, and tissue has to be repaired or reconstituted by activation of progenitors/stem cells (11,49). There are some similarities between these oval cells and our cultured cells multiplying in vitro. To assess for the progenitor cell phenotype in our proliferating cells, we used the OV6 antibody to visualize this accepted oval cell feature. This antibody is known to recognize the two different cytokeratins CK14 and CK19 (11). As the latter also detects bile duct cells in normal liver, anti-OV6 should be considered as an impartial ductular marker detecting progenitor as well as biliary features (51). In a similar sense, mature hepatocytes are devoid of CD49f, whereas it is strongly expressed on bile ducts as well as in progenitor cells including oval cells (12,18,45). We could also detect high expression of CD49f in our cultured hepatocytes, as well as the ductular markers CK7 and CK19. The loss of the functional marker Cx32 in our proliferating hepatocytes is also a known occurrence during the activation of oval cells (55).
Oval cells express a number of mesenchymal markers including vimentin, which is not expressed in mature hepatocytes (51). However, we could detect immunoreactivity to these filaments in our cultured cells, as well as vastly increasing mRNA levels during the course of culture. Vimentin filaments, expressed typically in cells of mesenchymal origin, are also expressed in neonatal hepatocytes, as well as fetal hepatic epithelial cells, being found concomitantly alongside cytokeratins such as CK18 (38). It is currently widely accepted that mesenchymal– epithelial transition plays a pivotal role in liver development and regeneration (8). Therefore, the coexpression of epithelial and mesenchymal markers in progenitor cells endorses our findings of coexpression in hepatocytes stimulated into proliferation in vitro.
The prevalence of CD133 expression in stem and progenitor cells in numerous tissues was described recently (4,42,43,47). This cell surface marker is further expressed in fetal liver as well as in hematopoietic stem cells in the bone marrow. CD133, widely considered as a stem cell marker, was detected in our proliferating cells from the first day onward and is also known to be expressed in oval cells (54). There is increasing evidence that CD133-positive cells may be isolated from diseased or regenerating liver (5). Some of these populations are associated with epithelial progenitors, others with contributing mesenchymal cell types (19).
Bipotent Differentiation of Cultured Cells Into Hepatocytes and Biliary Cells Following Transplantation
As recently demonstrated, mouse immortalized embryonic stem cells differentiate into hepatocytes and bile ducts when transferred into the adult liver (48), and here, newly formed cells may rescue mice from hepatic injury (53). Human embryonic stem cells can be differentiated in vitro into hepatocyte-like cells and are able to express liver-associated proteins (39). Fetal hepatoblasts display bipotential features and are the precursors of hepatocytes and biliary epithelial cells. In adults, progressive differentiation from orthotopic progenitor cells into hepatocytes and bile duct cells may also occur when the regenerative capacity of mature hepatocytes is massively impaired (13,41,44).
The simulated hepatic milieu in vitro is likely to exert a decisive effect on the switch of phenotype of cultured cells and may also prepare for the further bipotent proliferation and differentiation capacity in vivo. The application of the alkaloid retrorsine, as used here, is widely known to trigger acute effects such as biliary epithelial cell proliferation and slight reduplication of bile ducts of the recipient liver (21,22). Accordingly, reconstitution of bile ductules from transplanted cells was already observed in recipient livers following pretreatment with retrorsine (56). In this study, engraftment could be explained as a direct result of implanting biliary cells. However, most publications in this context describe transdifferentiation of hepatocytes into biliary cells following the application of specific toxins addressing the biliary system or following the means of bile duct ligation (25,28,52). Both settings suggest that the liver per se has the ability to generate or even create the demand for biliary cells in the context of severe injury.
In summary, our studies demonstrated that primary adult hepatocytes isolated from healthy liver regain their potential to proliferate grossly when stimulated appropriately in vitro. At the same time, these proliferating hepatocytes adopted precursor characteristics usually attributed to intrahepatic progenitor cells or hepatoblasts. The newly expressed marker profile and phenotypic switching very much resembled a progenitor cell entity, which is usually described as an “oval cell” in the literature.
The culture conditions provided by CM supplemented with LiCl proved to yield the greatest number of cells as well as the most marked change to the desired progenitor profile based on marker expression. Thus, only these cells were selected for the in vivo studies. Following transplantation into a rat liver repopulation model, these retrodifferentiated hepatocytes demonstrated their bidirectional redifferentiation potential into hepatocytes and bile duct cells. Of note, this described cell plasticity of somatic hepatocytes being modified in vitro as well as in vivo was achieved by the pure alteration of the biological microenvironment and no genetic or epigenetic manipulation of the primary cells as such. The enormous plasticity of adult hepatocytes as demonstrated may help to advance current protocols for translational research and the development of novel clinical therapies. To our knowledge, this is the first report on a two-step modification process/model (in vitro followed by in vivo) to achieve bidirectional differentiation plasticity in a single mature cell entity, in this case, the adult hepatocyte.
Footnotes
Acknowledgments
The authors would like to thank Sabine Wolfgramm and Sabrina Goldmann for their excellent work in preparing the immunolabeling figures and PCR analyses for this article. We express our gratitude to Andrew Entwistle and Gabriel Cantanhede for their critical review of the manuscript. The authors declare no conflicts of interest.