
Abstract
This study was designed to assess liver-specific functions of human fetal liver cells proposed as a potential source for hepatocyte transplantation. Fetal liver cells were isolated from livers of different gestational ages (16-22 weeks), and the functions of cell preparations were evaluated by establishing primary cultures. We observed that 20- to 22-week-gestation fetal liver cell cultures contained a predominance of cells with hepatocytic traits that did not divide in vitro but were functionally competent. Fetal hepatocytes performed liver-specific functions at levels comparable to those of their adult counterpart. Moreover, exposure to dexamethasone in combination with oncostatin M promptly induced further maturation of the cells through the acquisition of additional functions (i.e., ability to store glycogen and uptake of indocyanine green). In some cases, particularly in cultures obtained from fetuses of earlier gestational ages (16-18 weeks gestation), cells with mature hepatocytic traits proved to be sporadic, and the primary cultures were mainly populated by clusters of proliferating cells. Consequently, the values of liver-specific functions detected in these cultures were low. We observed that a low cell density culture system rapidly prompted loss of the mature hepatocytic phenotype with downregulations of all the liver-specific functions. We found that human fetal liver cells can be cryopreserved without significant loss of viability and function and evaluated up to 1 year in storage in liquid nitrogen. They might, therefore, be suitable for cell banking and allow for the transplantation of large numbers of cells, thus improving clinical outcomes. Overall, our results indicate that fetal hepatocytes could be used as a cell source for hepatocyte transplantation. Fetal liver cells have been used so far to treat end-stage liver disease. Additional studies are needed to include these cells in cell-based therapies aimed to treat liver failure and inborn errors of metabolism.
Introduction
Human adult hepatocyte transplantation to treat some liver-based inborn errors of metabolism and liver failure entered clinical practice approximately 20 years ago (28,41) as an alternative to orthotopic liver transplantation. The procedure was shown to improve and/or partially correct metabolic defects (9,14,35,38). However, it was also shown to offer no sustained benefits in the majority of cases (36). A major challenge of hepatocyte transplantation is the limited supply of donor organs for isolating relevant numbers of good quality cells (38). Hepatocytes for transplantation are obtained from rejected organs for transplantation, such as steatotic livers or those that have undergone a long cold ischemia time, but also from surgical leftovers after liver transplantation (29). Our group carried out a Phase I clinical study in which transplantation of freshly isolated human fetal liver cells (FLCs) was used to bridge patients with end-stage liver disease to liver transplantation. The isolation procedure of FLCs, including a case report, has been recently published (11). The procedure was found to be safe and also led to the improvement of clinical scores, thus confirming previous reports (21).
In this study, fetal hepatocytes are proposed as an alternative source of cells for transplantation. Our aim was to more precisely evaluate phenotype and liver-specific functions of the FLCs obtained after collagenase perfusion of a human fetal liver, and determine whether they can support impaired or lost hepatic functions. With the exception of a few reports (12,19—21), there is little documentation in the literature of transplantation of human FLCs in clinical practice. In addition, extensive characterization of human liver cells of fetal origin is reported in only a few studies (6,22,26), which mainly focus on hepatic stem cell characterization (22) and cell immunophenotype (6), rather than on liver-specific functions of fetal hepatocytes.
While the liver of adult organisms performs a multitude of functions related to metabolism and detoxification, the fetal liver is a transient site of hematopoiesis and serves as a primary source of red blood cells up to the 24th week of gestation, while defined hepatic functions are fully acquired near birth (16,18). Erythroid cells have been shown to reside within the fetal liver parenchyma from midgestation to the time of birth, and a function of the fetal liver as a niche for maturation of erythroid cells has been suggested (15). As cells expressing hepatic markers begin to appear in second trimester human fetal livers (32), a considerable number of cells populating the fetal liver parenchyma are expected to be hematopoietic. Typically, hepatic functions are tested in vitro on primary cultures of hepatocytes obtained after collagenase digestion of the liver. Hepatocytes are plated onto collagen- or other extracellular matrix protein-coated dishes and maintained in hormonally defined serum-free culture media (3,34). Because of the limited replicative capacity of the cells and rapid loss of the hepatocytic phenotype, culturing of hepatocytes has proven to be difficult (2). However, the in vitro assessment of hepatic functions does not require long-term culturing of the cells (5,10,34) because some liver-specific functions can be evaluated just a few hours after plating.
We focused our attention on the most representative hepatic functions, such as albumin secretion (a key marker of hepatic functioning), urea synthesis, and activity of cytochrome enzymes from the subfamily cytochrome P450 (CYP), family 3, subfamily A (CYP3A), particularly polypeptide 7 (CYP3A7) and 4 (CYP3A4) isoforms, which are known to be the most abundant cytochromes in the fetal and the adult liver, respectively. We also evaluated glucose 6-phosphatase (G6Pase) enzyme activity, glycogen storage, and uptake of the vital stain indocyanin green (ICG). We used commercially available human adult hepatocytes and human fetal liver mesenchymal stromal cells (FL-MSCs) as positive and negative controls, respectively. In addition to cell function, we investigated the proliferative capacity of fetal hepatocytes by culturing them in low cell density.
A critical issue during the transplantation of human FLCs is the limited number of hepatocytes obtained from a single donor fetus. Augmenting cell numbers would require pooling thawed cell suspensions from different donors to be transplanted in a single recipient, a strategy that is currently employed in adult hepatocyte transplant programs (30,31). In view of this, we tested the effects of cryopreservation on cell viability, ability to adhere to culture dishes, and cell function. If cryopreservation proves successful, it will allow for the banking of FLC suspensions to be used for programmed infusions of larger numbers of cells.
Materials and Methods
Tissue Procurement
Human fetal livers (Ospedale Civico, Palermo, Italy) were taken from 16 to 22 weeks gestation human fetuses obtained in cases of therapeutic abortions according to a protocol approved by ISMETT Institutional Research Review Board. Informed consent was obtained for each donor. Data on gestational age are shown in Table 1.
Gestational Age, Cell Yield, Viability, and Plating Efficiency of Human Fetal Samples

Flow Cytometry Analysis
To assess the immunophenotype of the different cell fractions in our FLC preparations, a flow cytometric analysis was performed as previously described (8). The primary antibodies (1:20) used were immunoglobulin G1-fluorescein isothiocaynate (IgG1-FITC), IgG2—phycoerythrin (PE), cluster of differentiation 45 (CD45)—FITC, CD34—PE, CD90—PE, glycophorin A (Gly-A)-PE Ki-67—FITC, CD106—allophycocyanine (APC), epithelial cell adhesion moleocule (EpCAM)—FITC (all BD Pharmingen, Franklin Lakes, NJ, USA), cytokeratin 19 (CK19)—PE (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), and CK18—FITC (Abcam, Cambridge, UK).
Cell Cultures and Media Composition
FLCs were isolated, as previously described (11), by collagenase (NB 1 GMP grade; Serva, Electrophoresis GmbH, Heidelberg, Germany) perfusion of human fetal livers. Data on cell yield and viability are shown in Table 1. FLCs were plated onto collagen I-coated six-well plates (BD Biosciences, Franklin Lakes, NJ, USA) in Kubota's medium (KM) (43) containing 5% fetal bovine serum (FBS; Lonza, Basel, Switzerland). After cell attachment (approximately 8 h), cultures were switched to serum-free medium. KM reagent sources were Roswell Park Memorial Institute 1640, glutamine (Lonza) and antibiotics (penicillin—streptomycin; Lonza), bovine serum albumin (BSA), nicotinamide, transferrin, hydrocortisone, 2-mercaptoethanol, selenium, zinc sulfate, oleic acid, linoleic acid, linolenic acid, and palmitic acid, palmitoleic acid (all from Sigma-Aldrich, St. Louis, MO, USA), and insulin (Humulin; Lilly, Indianapolis, IN, USA). Furthermore, we used two different culture systems: a high cell density (approximately 1.8 × 105 cells/cm2) and a low cell density system (approximately 0.6 × 105 cells/cm2). The low cell density system was used to evaluate the proliferative activity of FLCs.
We also cultured fetal hepatocytes in a differentiation medium (DM), which consisted of KM supplemented with dexamethasone (10−7 M; Hospira, Lake Forest, IL, USA) and oncostatin M (OSM; 10 ng/ml; R&D Systems, Minneapolis, MN, USA).
Isolation of Human Fetal Liver Mesenchymal Stromal Cells (FL-MSCs) From Primary Cultures of Fetal Hepatocytes
After 10—15 days of culture, fetal hepatocytes died, and spindle-shaped cells, termed FL-MSCs, were harvested by trypsinization (0.05% trypsin, 0.02% ethylenediaminetetraacetic acid; Lonza), and transferred to untreated six-well plastic dishes (Sarstedt, Numbrecht, Germany).
Immunofluorescence Analysis
For immunofluorescence staining, the cells were fixed according to the antibody manufacturer's instructions and processed as previously described (7). The primary antibodies used were anti-CK18, 1:100 (Abcam), anti-albumin, 1:500 (Sigma-Aldrich), anti-α 1-antitrypsin, 1:100 (Sigma-Aldrich), anti-α fetoprotein (AFP), 1:100 (Santa Cruz Biotechnologies), and anti-transferrin, 1:100 (Santa Cruz Biotechnologies). The cells were mounted with Prolong Gold Antifade (Invitrogen), including 4′,6-diamidino-2-phenylindole (DAPI) for the nuclear counterstaining and stored in the dark.
Immunocytochemistry Analysis
Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) and incubated with methanol/3% H2O2 (both from Sigma-Aldrich) for 30 min to deactivate the endogenous peroxidase. After blocking with phosphate-buffered saline (PBS; Lonza) 0.5% Tween 20 (Sigma-Aldrich) 3% BSA for 1 h, cells were incubated with the primary antibody anti-factor VIII 1:1,000 diluted (gift from Raffaella Gentile, ISMETT) overnight in a humid chamber at 4°C. The cells were then stained with the secondary antibody using the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA, USA) and incubated with 3,3′-diaminobenzidine (DAB) chromogen substrate using the DAB Substrate Kit for peroxidase (Vector Laboratories).
In Vitro Functional Analysis
Functional assays were carried out in both high- and low-density cultured cells. Cells were plated onto six-well collagen I-coated dishes and cultured in 1 ml of KM. Culture supernatants were collected 1 h, 6 h, and 24 h after cell attachment and used for albumin secretion and urea synthesis testing. For albumin secretion, samples were diluted 1:4 to fit the assay concentration range. CYP activity was performed 1 h after cell attachment. The remaining functional assays (G6Pase activity, glycogen storage, and ICG uptake) were carried out after 48 h of culturing. Plated human adult hepatocytes (Lonza) and cryopreserved human adult hepatocytes (Life Technologies, Carlsbad, CA, USA) were used as positive control cells, while the FL-MSCs were used as negative control cells.
Albumin Secretion and Urea Production
The secretion of albumin was measured using the enzyme-linked immunosorbent assay (ELISA) kit (Abnova, Heidelberg, Germany) according to manufacturer's instructions. The absorbance was measured on a microplate reader (Model 680; Bio-Rad, Hercules, CA, USA). Urea production was determined by a blood urea nitrogen (BUN) test using an automated system (Dimension RxL Max; Siemens Healthcare Diagnostics, Tarrytown, NY, USA). The results of both assays were normalized to the total number of attached cells and expressed as μg/ml/106 cells/h. Cells were detached by trypsinization and counted by trypan blue (Lonza) exclusion test.
Activity of Cytochrome P450 (CYP Assay)
Activities of isoforms −3A7 and −3A4 of CYP3A were assessed by using the P450-Glo Assay [luciferin-PFBE (6′-pentafluoro-benzyl ether) and luciferin-IPA (isopropyl acetal), respectively; Promega, Madison, WI, USA] according to manufacturer's instructions for nonlytic CYP. Cultured cells were incubated with medium supplemented with pGlo substrates. One hour (luciferin-IPA) and 4 h (luciferin-PFBE) after exposure, 25 μl of medium and 25 μl of luciferin detection reagent were added to a white opaque 96-well plate (Corning Inc., Corning, NY, USA) and read on a luminometer (Glomax; Promega) after 20-h incubation at room temperature. To determine the inducible activity of CYP3A4, cells were treated with the specific inducer rifampicin (10 μM for 48 h; Sigma-Aldrich) (27). The results were normalized to the total number of attached cells and expressed as relative light units (RLU)/106 cells/h.
Glucose-6-Phosphatase (G6Pase) Activity
The activity of the enzyme G6Pase was assessed according to methods described by Sokal et al. (39), with minor modifications. Briefly, cultured cells were incubated with Tris-acetate buffer pH 6.5 containing 2.08 mM glucose-6-phosphate and 2.4 mM nitric lead (all from Sigma-Aldrich) for 20 min at 37°C in a humidified atmosphere of 5% CO2. After rinsing in distilled water, cells were incubated with 1% ammonium sulfide (Sigma-Aldrich) for 15 s at room temperature to convert lead nitrate into brown lead sulfate and rinsed carefully with distilled water. The reaction was evaluated under an inverted CKX41 microscope (Olympus, Tokyo, Japan) coupled with a DS-Fi1 camera and Digital Sight (Nikon, Melville, NY, USA) for image acquisition.
Periodic Acid-Schiff (PAS) Stain for Glycogen Accumulation
Cultured cells were fixed with 4% paraformaldehyde for 10 min and incubated with 0.5% periodic acid (PA; Sigma-Aldrich) solution for 5 min at room temperature. After washing with distilled water, cells were covered with Schiff reagent (Sigma-Aldrich) for 15—20 min at room temperature, followed by three washes with tap water. Cells were examined under an inverted microscope, and image acquisition was done as described in the G6Pase activity subsection, above.
Indocyanine Green (ICG) Uptake Assay
Cultured cells were rinsed carefully with sterile PBS, the medium switched to prewarmed KM 1 mg/ml ICG (Cardiogreen; Sigma-Aldrich), and incubated for 15 min at 37°C, in the dark, in an incubator with 5% CO2. After rinsing with PBS, cells were observed under an inverted microscope, and image acquisition was done as described in the G6Pase activity subsection, above.
In Vitro Maturation of Fetal Hepatocytes: Dexamethasone and Oncostatin M Treatment
To induce maturation of fetal hepatocytes (17), high cell density plated cells were cultured for 6—10 days in DM. Maturation of fetal hepatocytes was evaluated by microscopic observation and by comparing gene expression and functional activities of undifferentiated and differentiated cells.
Gene Expression: Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA extraction, cDNA synthesis, and RT-PCR were performed as previously described (7). The primer sequences (Invitrogen, Carlsbad, CA, USA) are listed in Table 2.
Primers for RT-PCR
GAPDH, glyceraldehyde 3-phoshphate dehydrogenase; AFP, α fetoprotein; CYP3A4, cytochrome P450 family 3 subfamily A polypeptide 4; CK19, cytokeratin 19; HNF-4, hepatocyte nuclear factor 4.
Cryopreservation/Thawing of FLC Suspensions
The cryopreservation protocol for FLCs was based on the use of dimethyl sulfoxide (DMSO; Sigma-Aldrich), which has been shown to be the ideal cryoprotectant in most studies (40). William's E-based Heparmed medium (Biochrom, Berlin, Germany) was supplemented with insulin, transferrin, and glucagon (ITG; Biochrom), 30% FBS, and 10% DMSO. We used a freezing protocol with a freezing density of 5 × 106 cells/ml, as described by Mitry et al. (31), with minor modifications. Cells were stored at −80°C overnight and then in liquid nitrogen for long-term storage. For thawing, cells were removed from the liquid nitrogen tank and immediately placed in a water bath at 37°C. Cells were suspended in an ice-cold thawing solution (Heparmed 5% FBS) at 1:20 dilution and washed twice at 50 × g, 4°C for 5 min. Viability of thawed FLCs was assessed by the trypan blue exclusion method and compared to that of freshly isolated FLCs. Flow cytometry analysis was used to evaluate the immunophenotype of thawed FLCs. Thawed cells were also plated onto collagen I-coated six-well dishes and tested for their normal culture behavior and functions.
Plating Efficiency of FLCs
Owing to the predominance of erythroid GlyA+ cells in FLC suspensions, the calculation of plating efficiency was based on the number of CK18+ epithelial/hepatocytic cells, by counting the number of nonadherent CK18+ cells evaluated by flow cytometry. The result was expressed as a percentage of the initially plated CK18+ cells. We also evaluated the ability of thawed cells to form a compact hepatocytic monolayer.
Statistics
Data analyses were performed by using SigmaStat for Windows (Microsoft, Redmond, WA, USA). All data were expressed as mean ± SD. Data from two different groups were compared using the Student's t-test. Differences between the groups were considered significant at a value of p < 0.05.
Results
Cell Yield, Viability, and Plating Efficiency of Freshly Isolated FLCs
The mean viability of freshly isolated FLCs was 82.3 ± 3.9%, with a cell yield of 0.9 × 109 ± 0.39 cells, and a plating efficiency of 71.6 ± 12.9% (mean ± SD representing 13 FLC samples) (Table 1).
Immunophenotype of Freshly Isolated FLC Suspensions
We used early (16—19 weeks gestation; n = 6) and late (20—22 weeks gestation; n = 7) FLC samples. Significant differences between early and late FLCs were the high number of Ki-67+ proliferating cells and CK18+/ CK19+ cells observed in early groups. The percentage of Ki-67+ cells was 57.3 ± 4.6% in early versus 3.7 ± 4.7% in late FLCs (p < 0.001), while the CK18+/CK19+ cells were 15.6 ± 3.8% in early versus 4.17 ± 3.6% in late FLCs (p < 0.05) (Fig. 1A). A predominance of erythroid Gly-A+ cells (approximately 70%) was detected in FLC suspensions from both early and late groups, while the percentage of CK18+ epithelial/hepatocytic cells in both groups was approximately 30% (Fig. 1A). Cytospins from freshly isolated FLC suspensions and stained with anti-albumin also showed a small subset of positive hepatocyte cells (Fig. 1B). Early and late FLC suspensions consisted of comparable percentages of EpCAM+ stem/precursor cells (3.45 ± 0.35% in early and 5.6 ± 5.02% in late), CD34+ hematopoietic cells (1.85 ± 0.49% in early and 1.16 ± 1.2% in late), CD90+ mesenchymal cells (10.03 ± 8.2% in early and 4.1 ± 3.9% in late), and CD45+ leukocytic cells (5.25 ± 6.4% in early and 6.2 ± 4.3% in late). Furthermore, approximately 2% of cells from both groups were positive for the endothelial marker CD106 (data not shown).

Immunophenotype, morphology, and expression of liver-specific proteins in human FLCs. (A) Flow cytometry analysis of early versus late freshly isolated FLCs. Gly-A, glycophorin A; CK18, cytokeratin 18. (B) Immunofluorescence showing freshly isolated FLCs on cytospin stained with anti-albumin antibody (ALB; scale bar: 50 μm). (C) Spindle-shaped FL-MSCs in collagen I-coated dishes on day 15 of culture. (D) Twenty-one-week gestation fetal hepatocytes cultured in high cell density (0.6 × 105 cells/cm2) 2 days after plating onto a collagen I-coated dish. (E) Positive control, adult hepatocytes on day 2 of culture. Scale bar: 50 μm. (F) Cultured 21.5-weeks gestation fetal hepatocytes on chamber slides stained with anti-α-1-antitrypsin (ATT), anti-transferrin (TRA), and anti-albumin antibodies. Blue: DAPI (4′,6-diamidino-2-phenylindole) for nuclear counterstaining. Scale bar: 25 μm. Plotted values (mean ± SD) represent early (n = 6) and late (n = 7) samples, respectively. *p < 0.05; **p < 0.001; differences not denoted with an asterisk are not significant.
Assessment of Hepatic Functions in Cultured Cells
We used late FLCs (n = 7), adult hepatocytes (n = 6), and FL-MSCs (n = 3).
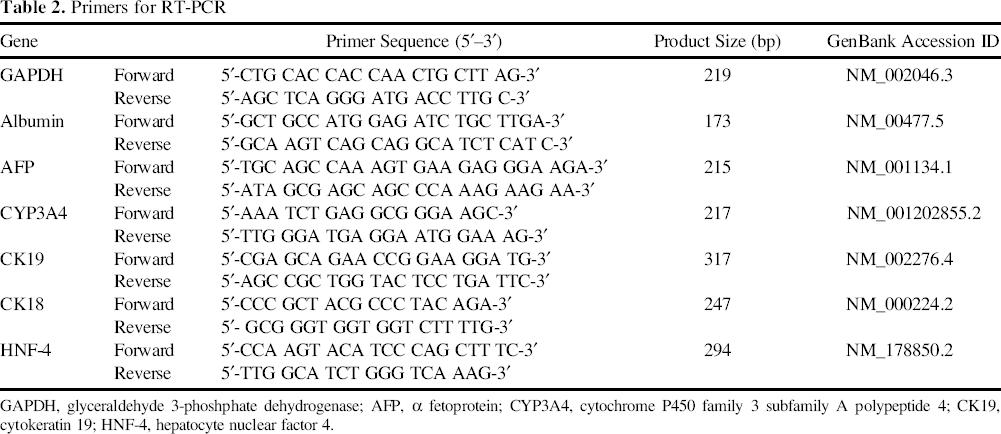
Isolated FL-MSCs (n = 3) presented a predominant mesenchymal morphology (Fig. 1C) and were positive for the mesenchymal marker CD90 (73.1 ± 3% positive). Adherent FLCs formed a compact hepatocyte monolayer with the typical intercellular connections visible as translucent spaces between cells (Fig. 1D), similar to the human adult hepatocyte control (Fig. 1E). Immunofluorescence staining showed that fetal hepatocytes were positive for liver-specific markers α-1-antitrypsin (ATT), transferrin (TRA), and albumin (ALB) (Fig. 1F). Fetal and adult hepatocytes secreted albumin into medium (0.13 ± 0.048 μg/ml/106 cells/h in fetal and 0.23 ± 0.039 μg/ml/106 cells/h in adult hepatocytes) (Fig. 2A), and synthesized urea (46.6 ± 11.5 μg/ml/106 cells/h in fetal and 56.6 ± 20.8 μg/ml/106 cells/h in adult hepatocytes) (Fig. 2B), while FL-MSCs did not secrete albumin, nor synthesize urea (Fig. 2A, B). Hepatocytes also expressed G6Pase enzyme activity, as indicated by positive cells stained brown as detected in fetal (Fig. 2C) and adult hepatocytes (Fig. 2D), but not in FL-MSCs (small panel). Fetal hepatocytes showed a higher activity of CYP3A7 compared to adult hepatocytes (1,099,731 ± 103,209 RLU/106 cells/h in fetal and 33,788 ± 18,205 RLU/106 cells/h in adult hepatocytes; p < 0.0001) (Fig. 2E). The activity of CYP3A4 was lower in fetal compared to adult hepatocytes (49,132 ± 11,039 RLU/106 cells/h in fetal and 248,311 ± 36,604 RLU/106 cells/h in adult cells) (Fig. 2F). Exposure to rifampicin determined a strong induction of CYP3A4 activity in adult hepatocytes (596,789 ± 62,618 RLU/106 cells/h; p < 0.0001), and a nonsignificant induction in fetal hepatocytes (Fig. 2F). FL-MSCs did not show any CYP activities (Fig. 2E, F).
Assessment of the hepatic function in cultured fetal and adult hepatocytes and FL-MSCs. (A) Albumin secretion measured by ELISA in culture supernatant at 1 h, 6 h, and 24 h after cell attachment. FET, fetal hepatocytes; AD, adult hepatocytes. (B) Urea synthesis measured by BUN test in culture supernatant at 1 h, 6 h, and 24 h after cell attachment. (C) Activity of glucose 6-phosphatase (G6Pase) enzyme assessed by cytochemistry. (D) Positive control for G6Pase activity, adult hepatocytes, and in the small panel, negative control FL-MSCs. (E) Activity of cytochrome P450 family 3 subfamily A polypeptide 7 (CYP3A7) assessed with the P450-Glo assay (luciferin-PFBE). (F) Basal and rifampicin-induced activity of CYP3A4 assessed with the P450-Glo assay (luciferin-IPA). The results were normalized to the total number of attached cells. Cells were detached by trypsinization and counted by trypan blue exclusion test. For albumin and urea assays, plotted values (mean ± SD) represent fetal hepatocytes (n = 7), and adult hepatocytes (n = 6). For CYP assays, plotted values (mean ± SD) represent fetal hepatocytes (n = 4), and adult hepatocytes (n = 3). *p < 0.05; ***p < 0.0001; differences not denoted with an asterisk are not significant.
Dexamethasone/OSM-Induced Maturation of Fetal Hepatocytes
We compared undifferentiated with differentiated fetal hepatocytes (n = 3). Fetal hepatocytes plated at high cell density and cultured in DM acquired additional hepatic functions. Unlike undifferentiated fetal hepatocytes, which did not show glycogen storage (Fig. 3A) and ICG uptake (Fig. 3B), fetal hepatocytes in DM accumulated glycogen (Fig. 3C), and showed ICG uptake (Fig. 3D). The in vitro maturation was also accompanied by changes in gene expression, such as reduction in the early hepatic marker AFP, abolishment of the hepatic precursor marker CK19, and a slight increase in mature hepatic markers CYP3A4, and CK18 mRNAs, while HNF-4 and albumin mRNAs were practically unchanged (Fig. 3E).

In vitro maturation of fetal hepatocytes. (A) Undifferentiated 21.5-week gestation fetal hepatocytes stained with PAS. (B) ICG uptake assay in undifferentiated 21.5-week gestation fetal hepatocytes. The same sample grown in DM and stained with PAS (C), and with ICG (D). Scale bar: 50 μm (A—C); 25 μm (D). (E) Representative RT-PCR for gene expression analysis in undifferentiated and differentiated fetal hepatocytes. U, undifferentiated; D, differentiated. AFP, α fetoprotein; HNF-4, hepatocyte nuclear factor 4.
Hepatic Morphology and Function in Low Cell Density Cultures
When fetal hepatocytes were cultured at low cell density (n = 3) (Fig. 4A), no direct hepatocyte proliferation was observed, but scattered flat cells appeared on days 4—6 (Fig. 4B) and became confluent by day 10, while the hepatocytic cell type disappeared (Fig. 4C). Furthermore, hepatic functions, such as albumin secretion and CYP3A7 activity, were significantly downregulated in flat cell cultures (albumin secretion: 0.028 ± 0.013 μg/ml per 106 cells/h; p < 0.001; CYP3A7 activity: 270.888 ± 139.388 RLU/106 cells/h; p < 0.0001) (Fig. 4D).
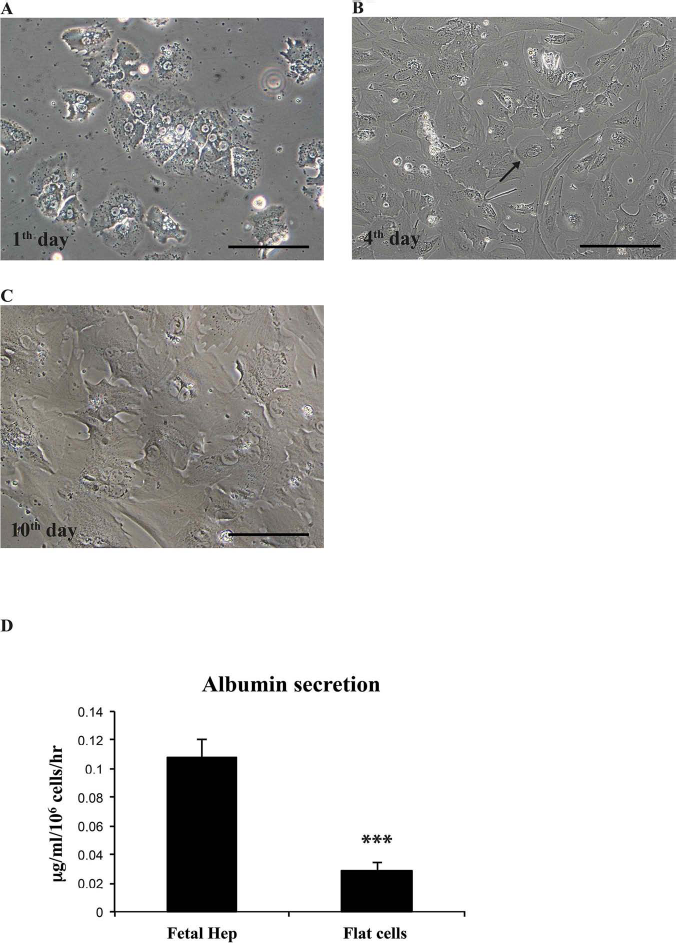
Low cell density cultures of fetal hepatocytes. (A) Low cell density culture on day 1. (B) Presence of flat cells, indicated with a black arrow, on day 4 of culture. (C) Confluent flat cells on day 10 of culture. Scale bar: 25 μm (A); 50 μm (B); 25 μm (C). (D) Albumin secretion in fetal hepatocytes and flat cell cultures (0.10 ± 0.013 μg/ml/106 cells/h in fetal hepatocytes, and 0.028 ± 0.006 μg/ml/106 cells/h in flat cells. Plotted values (mean ± SD) represent fetal hepatocytes (n = 3) and flat cells (n = 3). **p < 0.001.
Early FLC Cultures
In early FLC cultures (n = 6), at least two main types of spherical cell colonies were distinguishable. The cells in one colony were morphologically similar to hepatocytes, but smaller in size (Fig. 5A, black arrow). The cells in the other colony had epithelial traits, but were morphologically different from hepatocytes (Fig. 5A, white arrow). These epithelial cells were negative for albumin (Fig. 5B) and positive for AFP (Fig. 5C) and CK18 (Fig. 5D). Both cell colonies proliferated within a few days (Fig. 5E, F). Additional detected cells were factor VIII+ cells, observed on day 6 of culture (Fig. 5G), and factor VIII+ capillary-like sprouts observed on day 25 (Fig. 5H). However, the cell colonies observed in these early cultures never became confluent, and by days 25—28 the early FLC cultures were populated exclusively by cells with a mesenchymal appearance (Fig. 5I).

Eighteen-week gestation FLCs in culture. (A) Two different cell colonies indicated with black arrow (hepatoblast-like) and white arrow (epithelial-like) detected on day 6 of culturing. Epithelial-like colony stained with anti-albumin (B), anti-AFP (C), and anti-CK18 antibodies (D). (E) Proliferating hepatoblast-like colony on day 15 of culturing. (F) Proliferating epithelial-like cells on day 15 of culturing. (G) Single cells observed on day 6 and stained with anti-factor VIII antibody. (H) Capillary-like sprout observed on day 25 and stained with anti-factor VIII antibody. (I) Predominance of the fibroblastic cells on day 25 of culturing. Blue: DAPI for nuclear counterstaining. Scale bar: 50 μm (A—F, I); 25 μm (G, H).
Cryopreservation/Thawing of FLCs
Assessment of viability and flow cyotmetry analysis were applied to freshly isolated (n = 8) and thawed (n = 8) FLCs. The mean percentage of viability of thawed FLC suspensions evaluated up to 1 year of storage was 73 ± 4.1% (Fig. 6A). We observed a twofold reduction in percentage of Gly-A+ cells (61.3 ± 14.98% in fresh vs. 32.9 ± 16.9% in thawed cell suspensions; p < 0.05), which was accompanied by an increase in CK18+ cells (24.4 ± 8.5% in fresh and 53.6 ± 15.06% in thawed cell suspensions; p < 0.05), and in EpCAM+ cells (6.8 ± 5.5% in fresh and 15.8 ± 4.5% in thawed cell suspensions; p < 0.05) (Fig. 6B). Furthermore, we detected a reduction in CK18+/CK19+ cells that was not significant in late FLCs, but was significant in early FLCs (14.8 ± 5.2 in fresh and 3.8 ± 3.6 in thawed cell suspensions; p < 0.05). We did not observe significant changes in percentage of cells expressing the other markers (CD34+, CD45+, and CD90+). Albumin secretion (Fig. 6C), urea synthesis (Fig. 6D), and CYP3A7 activity (Fig. 6E) were exhibited by thawed fetal hepatocytes (n = 3) at levels comparable to those of fresh hepatocytes (n = 3).

Immunophenotype and functional evaluation of freshly isolated FLCs compared with thawed cells. (A) Viability of freshly isolated (n = 8) and thawed (n = 8) FLCs calculated by trypan blue exclusion assay. (B) Flow cytometric characterization of freshly isolated FLCs compared with thawed cells. (C) Albumin secretion, (D) urea synthesis, and (E) CYP3A7 activity in fresh (n = 3) and thawed (n = 3) FLCs. (F) Twenty-two-week gestation thawed FLCs after 1 year of storage in liquid nitrogen, plated onto a collagen I-coated dish on day 2. Scale bar: 100 μm. (G) Twenty-two-week gestation thawed FLCs after 20 months of storage in liquid nitrogen, plated onto a collagen I-coated dish on day 2. Scale bar: 25 μm. Plotted values (mean ± SD) represent fresh (n = 8) and thawed (n = 8) FLC samples. *p < 0.05.
Plating Efficiency of Thawed FLCs
Plating efficiency analysis was applied to thawed FLCs (n = 8) and compared to that of the corresponding fresh cells. In FLCs thawed 10 months later, the number of CK18+ cells detected in culture supernatants collected 8—10 h after plating, which reflect the number of nonadherent CK18+ cells, was <13% and corresponded to 19—25% of the number of total CK18+ cells initially seeded. This yielded a plating efficiency of 68.3 ± 10.7% (Table 1), which was not statistically significant compared with that of freshly isolated cells. Thawed FLCs formed an intact monolayer, and microscope observations revealed no morphological differences compared with monolayers from fresh FLCs (Fig. 6F). Twenty months later, thawed FLCs showed a reduced plating efficiency (42.6 ± 5.5%), and some of the attached cells were large and flat and exhibited a tendency to lose hepatocytic morphology (Fig. 6G).
Discussion
In line with data in the literature (15), we found that cell suspensions obtained after collagenase digestion of human fetal livers contained approximately 35% CK18+ cells of hepatocytic/epithelial lineage, while the remaining percentage was represented mainly by Gly-A+ erythroid cells. How much of this erythroid contamination is actually due to fetal liver erythropoiesis or to inefficient organ washing/perfusion before collagenase digestion remains unclear.
We observed that the majority of cells observed in primary cultures of late (20-22 weeks gestation) FLCs were similar to adult hepatocytes in terms of morphology, including little, if any, capacity to proliferate in vitro. Albumin secretion, urea synthesis, and G6Pase activity were detected at approximately the same levels as those of control adult hepatocytes. Additional assessed functions as storage of glycogen and ICG uptake were easily acquired following in vitro maturation of cells. However, the PAS stain used here is not specific for glycogen because it stains several glycoproteins including glycogen.
Concerning liver cytochromes, the activity of CYP3A4 isoform was detected at negligible levels compared to CYP3A7 activity. It is known that CYP3A7 is the major CYP3A isoform of the fetal liver, while it is present at a lower level in the adult liver (23,24,37). Conversely, CYP3A4 is very low before birth but increases rapidly thereafter, reaching 50% of adult levels between 6 and 12 months of age (8).
Additional cells detected in FLC cultures were cells resembling small hepatocytes, epithelial cells similar to previously described hepatic precursors (21), both visible as clusters of proliferating cells, and factor VIII+ capillary-like sprouts, which were likely endothelial precursors. These cells were particularly abundant in early FLCs (16 to 19 weeks gestation), while cells with hepatocytic traits were quite rare. Consequently, liver-specific activities in early FLC cultures were almost undetected. Differences between “early” and “late” FLCs were also evidenced by flow cytometry and included significant higher numbers of CK18+/CK19+ cells, and Ki-67+ cells detected in cell suspension from early FLCs. We are aware that the differrences between “early” and “late” FLC groups are only based on observations and need confirmation in more extensive studies before concluding that they might be related to fetal liver development.
On the basis of the previous considerations, we assume that the different cell types detected in FLC suspensions may have the potential to restore liver functions: the fetal hepatocytes may well support the compromised liver functions in the same manner as adult hepatocytes do; the other cell types with proliferative capabilities may engraft and, eventually, generate new hepatocytes in vivo. The possibility that these cells can generate hepatocytes in vivo is intriguing. To shed light on the relationship between precursor and mature hepatocytes, numerous studies have been conducted by infusing fetal liver cells in animal models (4,33,44,45).
We observed that the low cell density cultures accelerated the decline in hepatocytic traits, including downregulation of all assessed liver-specific functions. This is a known phenomenon, probably triggered by a disruption of the cell—cell contacts, which are known to stabilize the hepatocytic phenotype (1,25). Our observation supports the strategy of transplanting clusters of hepatocytes rather than single cells, which not only may favor organ engraftment but also preserve the function of transplanted hepatocyte.
The hepatocytic cells account for approximately 35% of FLC preparations, so it is questionable whether transplanting cell preparations from a single fetus is sufficient to sustain the lost functions of a diseased liver. We could increase cell numbers by pooling cell suspensions from different donors to be infused in a single recipient. Cryopreservation allows cell preparations to be stored, thus ensuring a sufficient cell supply for treatments. However, cryopreservation is known to affect viability and function of hepatocytes on thawing, especially the plating efficiency, which is a key factor for a proper engraftment of the target organ (13). Through preliminary experiments, we observed that cryopreservation had only a minor impact on FLC viability and function. The mean percentage of viability of thawed cells stored up to 1 year in liquid nitrogen was approximately 70%, and the attachment efficiency, as well as the tested hepatic functions, was not impaired. Where significant alterations in attachment efficiency were observed (in two cases, data not shown), both cell preparations were obtained from suboptimal human fetal livers, thus confirming that quality of fresh hepatocytes before cryopreservation, and conditions of the liver tissue from which hepatocytes are isolated, are also crucial in determining the quality of thawed cells (42).
Since cryopreservation resulted in a twofold reduction in the erythroid cell fraction, we may have found a double advantage in cryopreserving FLCs, as the procedure may ultimately increase the purity of cell preparations. Because of the difference in the percentage of CK18+/ CK19+ cells between the early and the late fresh FLC suspensions (Fig. 1A, C), it is difficult to evaluate the real impact of cryopreservation on bipotent cell viability, though our data indicate that we may lose part of these cells on thawing. Our studies of viability and function of thawed FLCs are preliminary and need to be continued for extended periods of time.
To the best of our knowledge, this study is the first approach to estimating functional competencies of early versus late second trimester and fresh versus thawed cultured human FLCs. Our findings indicate human fetal liver-derived cells as a valid alternative to adult hepatocyte transplantation in liver cell-based therapies and suggest a novel clinical approach for human FLC transplantation by introducing the use of thawed cells. This strategy may allow for an increase in the number of transplanted cells per recipient, which may, in turn, ultimately improve clinical outcomes.
Footnotes
Acknowledgments
The authors thank Mariangela Di Bella, Daniele Galvagno, Maria Grupillo, and Monica Miele for their invaluable support during the fetal liver cell isolation process. The authors are grateful to Salvatore Pasqua for his technical assistance, Raffaella Gentile of ISMETT's Pathology Laboratory for kindly providing the anti-factor VIII antibody, and Warren Blumberg of ISMETT's Language Services Department for his excellent language revision of the manuscript. The authors also thank Stephen Strom for his suggestions concerning CYP3A activity studies, Giampiero La Rocca for providing the G6Pase activity assay protocol, and the Gynecology Unit of the Ospedale Civico (Palermo, Italy) for providing the human fetuses. This study was supported by a grant from the Fondazione Ri.MED and from the University of Pittsburgh Medical Center. The authors declare no conflicts of interest.