
Abstract
We have previously demonstrated in acute myocardial infarctions that human umbilical cord blood mononuclear cells (HUCBCs), which contain hematopoietic, endothelial, and mesenchymal stem cells, reduce acute myocardial infarction size by ≥50% and preserve LV contractility. We hypothesize that the beneficial effects of HUCBCs are due to secretion of biologically active factors that activate in cardiac endothelial cells and myocytes the cell survival protein Akt. We determined by protein microarrays the growth factors and anti-inflammatory cytokines secreted by HUCBCs into culture media during 12 h of hypoxia (1% O2). We then determined by Western blots the effects of cell-free media from hypoxic-conditioned HUCBCs (HUCM) on activation of the cell survival protein Akt in human coronary artery endothelial cells and cardiac myocytes in culture during 24 h of 1% O2. We also determined in separate experiments endothelial cell and myocyte apoptosis by caspase-3 and Annexin V. In the present experiments, HUCBCs secreted multiple growth factors, anti-inflammatory cytokines, and inhibitors of metalloproteinase during normoxia and hypoxia. Human cord blood cells increased the concentration in culture media of angiopoietin, hepatocyte growth factor, interleukin-4, insulin-like growth factor, placental growth factor, vascular endothelial cell growth factor, angiogenin, and stem cell factor by 100 to >10,000% during 12 h of 1% O2 (p < 0.001). HUCM, which contained these biological factors, significantly increased Akt phosphorylation/activation in coronary artery endothelial cells and cardiac myocytes subjected to 24 h of 1% O2 by more than 60% (p < 0.05) and increased the antiapoptotic protein Bcl-2 expression by 34–50% in comparison with endothelial cells and myocytes treated without HUCM in 1% O2 (p < 0.05). HUCM also significantly decreased caspase-3 activity and decreased hypoxic endothelial cell and cardiac myocyte apoptosis by more than 40% in comparison with cells cultured without HUCM (p < 0.05). Inhibition of Akt activation in endothelial cells and myocytes by the sensitive and specific antagonist API-1 during 24 h of hypoxia nearly completely prevented the beneficial effects of HUCM on inhibiting caspase-3 activity and apoptosis. We conclude that HUCBCs secrete biologically active factors during hypoxia that activate survival proteins in endothelial cells and myocytes that significantly limit apoptosis.
Keywords
Introduction
Acute myocardial infarction (AMI) is the leading cause of morbidity and mortality in the western world and, according to the World Health Organization, will be the major cause of death in the world by the year 2020 (25).
Injured and infarcted myocardium due to coronary artery occlusion is a potent trigger for the myocardial expression of the inflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and IL-6 (8,10,31). TNF-α activates IL-1 and IL-6 and orchestrates the host tissue response to acute myocardial injury. Within the first hours after infarction, TNF-α and IL-1 can increase 15- to 50-fold in myocardial infarcts (10). These inflammatory cytokines are acutely expressed by all nucleated cell types in the myocardium and have the ability to self-amplify through positive feedback loops (8,10). In addition, these cytokines trigger additional inflammatory cytokine responses, generate free oxygen radicals, activate matrix metalloproteinases, and acutely attract macrophages, neutrophils, and lymphocytes to infarcted myocardium that mediate additional injury to cardiac myocytes and vascular endothelial cells.
Myocardial inflammatory cytokines and inflammatory neutrophils and T lymphocytes cause cytochrome C release from endothelial cell and myocyte mitochondria, caspase proteolytic enzyme activation, and apoptosis due to enhanced expression of the apoptotic proteins β-cell chronic lymphocytic leukemia (CLL)/lymphoma 2 (BCL2)-associated agonist of cell death (Bad) and BCL2-associated X (Bax) (19,26). In this regard, extensive apoptosis occurs during the initial 4–6 h after coronary occlusion and exceeds necrotic cell death by sixfold (20).
We have demonstrated that human umbilical cord blood mononuclear cells (HUCBCs), which contain hematopoietic, endothelial, and mesenchymal stem cells, can suppress in acutely infarcted myocardial tissue the expression of the inflammatory cytokines TNF-α, monocyte chemoattraction protein (MCP), macrophage inflammatory protein (MIP), and interferon-γ (INF-γ) by as much as 50% at 2–72 h after coronary artery occlusion (17). Human cord blood mononuclear cells also can limit within 12–72 h of acute coronary occlusion the myocardial infiltration of inflammatory neutrophils and lymphocytes into myocardial infarctions (17). In addition, we have demonstrated that HUCBCs cocultured with myocytes and coronary endothelial cells during 24 h of severe hypoxia (1% oxygen) can significantly limit endothelial cell and myocyte caspase expression and apoptosis (16).
The acute beneficial effects of HUCBCs in the treatment of acute myocardial infarctions that we have demonstrated (12–17) are unlikely to be due to cord blood cell transdifferentiation to coronary endothelial cells or cardiac myocytes. We hypothesize that HUCBCs secrete growth factors and anti-inflammatory cytokines that significantly limit or inhibit myocardial inflammatory cytokines and inflammatory cells and, in this manner, reduce myocardial apoptosis and infarct size. Therefore, in the present experiments, we determined the growth factors and anti-inflammatory cytokines secreted into cell culture media by HUCBCs during normoxia and during severe hypoxia. We then determined the effects of the cell-free media from the hypoxic-conditioned HUCBCs on the activation of the cell survival protein Akt (v-Akt murine thymoma viral oncogene homolog 1; protein kinase B) and on cell apoptosis and caspase activity in human coronary artery endothelial cells and cardiac myocytes during 24 h of hypoxia. The present investigation demonstrates that HUCBCs secrete biologically active factors that activate in coronary artery endothelial cells and cardiac myocytes the cell survival protein Akt that significantly reduces coronary endothelial cell and cardiac myocyte apoptosis during severe hypoxia.
Materials and Methods
The studies performed were approved by the Institutional Review Boards of the University of South Florida and the James A. Haley Medical Center.
HUCBC Cultures
Male and female human umbilical cord blood mononuclear cells were obtained from several local human cord banks (Saneron CCEL Therapeutics, Inc., Tampa, FL, USA; CryoCell Oldsmar, FL, USA; CordUse Orlando, FL, USA) and stored at −196°C in liquid nitrogen. The cord blood was rejected if the maternal blood was positive for human immunodeficiency virus, human T-lymphocytic virus, hepatitis, syphilis, or cytomegalovirus. The cryopreserved HUCBCs were thawed at 37°C and transferred into centrifuge tubes (Fisher Scientific, Fairlawn, NJ, USA) containing Isolyte S, pH 7.4 (Braun Medical, Melsungen, Germany). The cells were washed, centrifuged at 453 × g (1,500 rpm; swing bucket rotor A-4-62 for Eppendorf 5810R; New York, NY, USA), and the HUCBC viability was determined by Trypan Blue (Life Technologies, Grand Island, NY, USA) dye exclusion technique. HUCBC viability from each vial was greater than 90%. The HUCBCs contain approximately 1–2% CD34+ cells and 0.5% CD105+ cells as determined by fluorescent antibodies to CD34 and CD105 (SH2) cell antigens, which were obtained from Invitrogen, Carlsbad, CA, USA and Osiris, Columbia, MD, USA, and fluorescent-activated cell sorting cytometry (Becton Dickinson, Franklin Lakes, NJ, USA) in our facility. The remaining components of HUCBCs were predominantly hematopoietic precursor cells. The HUCBCs were not propagated in culture flasks.
HUCBCs in short-term cultures were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) with fetal bovine serum (Thermo Scientific, Waltham, MA, USA) and penicillin–streptomycin (Fisher, Franklin Lakes, NJ, USA) in T25 polyethylene tissue culture flasks (BD Biosciences, Franklin Lakes, NJ, USA). The HUCBCs in culture consisted of adherent large cells with big nuclei and floating small round cells with large nuclei that occupied the majority of the cytoplasm. In the absence of specific growth factors, the HUCBCs grow slowly in culture and do not propagate in ≤3 days.
Preparation of HUCBC-Conditioned Medium (HUCM)
Six million HUCBCs were plated with DMEM with 10% fetal bovine serum in T25 flasks (Fisher Scientific) in 10 experiments. The following day, the HUCBC cultures were serum starved with basal culture media without fetal bovine serum and subjected to hypoxia (1% O2, 94% N2, and 5% CO2). After 12 h, the medium without cells was collected and labeled as HUCBC-conditioned medium (HUCM). The HUCM was filtered with a 0.22-μm cellulose syringe filter (EMD Millipore, Billerica, MA, USA) and subjected to protein microarray analysis. The HUCM was used to treat coronary artery endothelial cells and cardiomyocytes that were subjected to 24 h of hypoxia (1% O2) in order to determine the effects of HUCM on hypoxic-induced endothelial cell and cardiomyocyte apoptosis. HUCM was also added to coronary artery endothelial cells or cardiomyocytes, and the cells were subjected to 24 h of normoxia (21% O2).
Protein Microarrays
Protein microarray experiments were performed from HUCBCs in normoxia (n = 5 experiments) or 1% oxygen (n = 10 experiments). Two milliliters of HUCM from each experiment was added to 2 ml of blocking buffer (RayBiotech, Atlanta, GA, USA). The solution from each experiment was placed on a separate protein array membrane (RayBiotech) in a plastic tray and incubated on a rocker plate for 2 h at room temperature (24). The solution was then aspirated, and each membrane was incubated on a rocker plate for 12 h at 4°C with a customized mixture of biotinylated primary antibodies, based on our preliminary experiments, that were specific for angiopoietin, hepatocyte growth factor (HGF), insulin-like growth factor (IGF), placental growth factor (PLGF), vascular endothelial growth factor (VEGF), angiogenin, IL-4, IL-10, stromal-derived factor (SDF), tissue inhibitor of metalloproteinase (TIMP), MCP, and fractalkine. The membranes were rinsed, and horseradish peroxidase (HRP)-conjugated streptavidin was added to each membrane. The membranes were then incubated at room temperature for 2 h, subjected to enhanced chemiluminescence detection for 60–120 s, and exposed on radiographic film (Amersham, Pittsburgh, PA, USA) for 90 s. The blots on the radiographic film were then scanned, and the protein density expression level was determined with ImagePro image analysis software (Media Cybernetics, Bethesda, MD, USA). All measurements were performed in duplicate, and the results were averaged for each measurement. The variation in duplicate determinations was less than 10% (17). The results for each growth factor or cytokine were then compared with and normalized to positive controls present on each membrane. In addition, Iscove's modified Dulbecco's culture medium (Life Technologies) was used as a normal control, and the proteins measured in this medium were subtracted from the proteins measured in the HUCM. The measurements during hypoxia were then expressed as a percentage of the normoxic measurements. Mean ± SEM values were then determined. The protein microarrays can detect as little as 10 pg/ml of growth factor or cytokine protein (17,24).
Human Coronary Artery Endothelial and Myocyte Cultures
Male human coronary artery endothelial cells were obtained from Lonza (CC285, Allendale, NJ, USA). Endothelial cells were maintained in endothelial growth media (Lonza) with fetal bovine serum (Lonza) and gentamycin (Gibco, Grand Island, NY, USA) and grown to approximately 80% confluence in T25 polyethylene tissue culture flasks (BD Biosciences, Pittsburgh, PA, USA).
Rat cardiac myocytes were obtained from ATCC (CRL-1446™, Manassas, VA, USA). The myocytes have morphological characteristics similar to embryonic cardiac myocytes but have hormonal, enzymatic, and electrical signal pathways similar or identical to adult cardiac myocytes (18,39). The cardiac myocytes were maintained in DMEM with fetal bovine serum and penicillin– streptomycin and grown to approximately 80% confluence in T25 polyethylene tissue culture flasks (BD Biosciences, Franklin Lakes, NJ, USA).
Coronary artery endothelial cell cultures (n ≥ 4 experiments) and cardiomyocyte cultures (n ≥ 3 experiments) were treated with either media taken from HUCBC cultures subjected to 12 h of hypoxia (1% O2, 94% N2, and 5% CO2) or normal culture media, and the endothelial cell and myocyte cultures were then subjected to 24 h of hypoxia or normoxia (21% O2). We have previously established that the oxygen saturation in infarcted myocardium averages 1–4%.
Western Blot Analyses for AKT and BCL-2
Coronary vascular endothelial cells or cardiac myocytes cultured in normoxia and hypoxia without or with HUCM were lysed with RIPA buffer (Sigma Aldrich, St. Louis, MO, USA) containing protease and phosphatases inhibitors (Pierce, Waltham, MA, USA). Protein determinations were performed with bicinchoninic acid protein kits (Pierce) with bovine serum albumin as the standard. Western blots were then performed for the survival protein Akt and for phospho-Akt, which is the activated form of Akt. Thirty micrograms of protein from cells from each culture was loaded and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad, Hercules, CA, USA) and transferred onto nitrocellulose membranes (EMD Millipore). The membranes were then blocked for 1 h in Tris-buffered saline (Bio-Rad) with Tween-20 buffer (Fisher Scientific) containing 5% nonfat milk (Bio-Rad). Immunoblotting was performed by incubating the membranes with monoclonal antibodies to Akt (1:1,000), p-Akt (Ser 473) (1:1,000), or Bcl-2 (1:1,000) (Cell Signaling, Beverly, MA, USA) in 5% nonfat milk overnight at 4°C and then with secondary antibodies (1:2,000; Santa Cruz Biotechnology, Dallas, TX, USA) conjugated with horseradish peroxidase for 3 h at room temperature. Actin (Invitrogen) was used as a standard control. After washing, the protein bands were detected by Supersignal West Pico chemiluminenscence kit (Thermo Scientific). The results were quantified with computer software (Image J, NIH, Bethesda, MD, USA).
Apoptosis Detection
Coronary vascular endothelial cells or cardiac myocytes were subjected to hypoxia or normoxia without or with HUCM for 24 h. After exposure, the cells were harvested through trypsinization (Life Technologies) and washed twice with cold PBS (0.15 M, pH 7.4). The cells were centrifuged at 453 × g (1,500 swing bucket rotor A-4-62 for Eppendorf 5810R) for 5 min, and the supernatants were discarded. The percentage of apoptotic cells was assessed based on the fluorescein isothiocyanate (FITC) Annexin V-binding assay (Invitrogen) (16). Briefly, the harvested cell pellets were resuspended in 1× binding buffer at a density of 1 × l06 cells per ml. One hundred microliters of the sample solution was transferred to a 5-ml culture tube (BD Biosciences, San Jose, CA, USA) and incubated with 5 μl of FITC-conjugated Annexin V for 15 min at room temperature in the dark. The cells were washed with binding buffer three times and then resuspended in 100 μl of binding buffer. Ten microliters was then deposited onto each slide. Cell nuclei were stained with 4′,6′-diamino-2-phenylindole·2HCl (DAPI; Life Technologies). ProLong Gold antifade reagent (P36930 Invitrogen, Carlsbad, CA, USA) was used for mounting media. Cells from each experiment were examined with fluorescence microscopy with FITC filters (Olympus, Central Valley, PA, USA) for Annexin V staining that is indicative of cell apoptosis. Four separate fields on each slide were randomly chosen that contained a minimum of 200 endothelial cells/field or 200 myocytes/field. Only Annexin V-positive cells with DAPI-stained nuclei were counted in order to avoid counting the endothelial cell or myocyte fragments. The results of the cell counts from the four separate fields on each slide were averaged.
In five separate experiments, flow cytometry analyses of 50,000 endothelial cells or myocytes/intervention were performed to determine apoptosis. The samples were analyzed by an Accuri flow cytometer (BD Bioscience). In these experiments, unstained endothelial cells and myocytes or endothelial cells and myocytes stained with Annexin V were analyzed.
Caspase Determination
Coronary artery endothelial cell (n ≥ 5 experiments) or cardiomyocytes (n ≥ 5 experiments) in cultures were serum starved for 24 h without or with the sensitive and specific Akt inhibitor API-1 [12.5 μM; NCI/Developmental Therapeutics Program (DTP) Open Chemical Repository, National Institutes of Health, Bethesda, MD, USA]. The cells were then subjected to incubation in normoxia or hypoxia for an additional 24 h without or with HUCM. Caspase-3 activity was measured using the caspase-3 colorimetric activity assay (Invitrogen) (9). Coronary endothelial cell or myocyte lysate was collected. The protein concentrations were measured using BCA protein assay kits (Pierce Biotechnology). The assay was based on hydrolysis of acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) by caspase-3, resulting in the release of p-nitroaniline (pNA) moiety (PromoKine, Heidelberg, Germany). p-Nitroaniline was detected at 405 nm (BioTek plate reader, BioTek, Winooski, VT, USA). Caspase-3 activity was calculated using a p-nitroaniline calibration curve as micromolar per gram of wet tissue. Caspase-positive controls (recombinant caspase-3) and negative controls with caspase-3 inhibitor were incorporated in the assay. The data were expressed as caspase U/μg endothelial cell or myocyte protein. One caspase unit is the amount of enzyme that will cleave 1.0 μmol of the substrate Ac-DEVD-pNA per minute at pH 7.4 at 25°C.
AKT Inhibition
The normoxia or hypoxia protocols with endothelial cells or myocytes without or with HUCM were repeated with the sensitive and specific Akt inhibitor API-1 (3,21,38). API-1 is a tricyclic nucleoside which, once inside cells, is phosphorylated to its monophosphate derivative by adenosine kinase. API-1 inhibits phosphorylation of Akt by binding to the PH domain of Akt, preventing phosphorylation at T308 and S473 and blocking its recruitment to the plasma membrane. API does not inhibit Ras-dependent extracellular signal-regulated kinase (ERK)1/2, mitogen-activated protein kinase, c-Jun N-terminal kinase (JNK), protein kinase C, serine/threonine-protein kinase (SGK), or protein kinase A (3,21,38). Separate vascular endothelial cell cultures (n ≥ 5 separate experiments) and cardiac myocyte cultures (n ≥ 3–5 separate experiments) without or with HUCM were then treated with the API-1 inhibitor and then subjected to either normoxia or hypoxia.
Data Analysis
All values were expressed as means ± SEM. The significance of the differences between values was assessed by Student's t test. When multiple comparisons among groups were performed, repeated measures analyses of variance were performed. Thereafter, the Bonferroni modification of the t test was used for planned comparisons, and Tukey's procedure was used for post hoc comparisons. Statistical significance was assigned when p < 0.05.
Results
HUCBCs Secrete Growth Factors and Anti-inflammatory Cytokines Into Media
Microarray protein determinations of the cell-free media from HUCBC cultures during normoxia demonstrated that HUCBCs secrete growth factors, anti-inflammatory cytokines, and inhibitors of metalloproteinases. Moreover, during 12 h of hypoxia, the HUCBCs increased by more than 100% the secretion of angiopoietin, HGF, anti-inflammatory IL-4, IGF, PLGF, VEGF, angiogenin, and SCF in comparison with these factors in normoxia (p < 0.001) (Fig. 1). Cord blood cells during hypoxia increased by ≥50% the secretion of fractalkine, IL-10, TIMP, and SDF (p < 0.01). The HUCBCs tolerated the 12 h of hypoxia well with less than 5% cell death due to loss of differentiated cells as determined by trypan blue staining. In this regard, hematopoietic, endothelial, and mesenchymal stem cells tolerate hypoxia well, and hypoxia maintains the pluripotency of stem cells (29). Human umbilical cord blood cell-free media after 12 h of hypoxia were added to coronary artery endothelial cells or cardiac myocytes, which were then subjected to 24 h of hypoxia.

Human umbilical cord blood cell (HUCBC) release of biological factors during 12 h of 1% oxygen. Each biological factor during hypoxia is expressed as percent increase above the value measured during normoxia. Mean ± SEM is presented. HGF, hepatocyte growth factor; IL-4, interleukin-4; IGF, insulin-like growth factor; PLGF, placental growth factor; VEGF, vascular endothelial cell growth factor; SCF, stem cell factor; TIMP, tissue inhibitor of metalloproteinase; SDF, stromal derived factor.
HUCBC-Conditioned Media Activate Akt in Coronary Endothelial Cells and Cardiac Myocytes
Coronary artery endothelial cells were subjected to 24 h of hypoxia without or with HUCM or 24 h of normoxia. Hypoxia significantly decreased activated p-Akt in endothelial cells cultured without HUCM in comparison with endothelial cells in normoxia (Fig. 2A, B). However, Akt and actin did not significantly change in either the normoxic or the hypoxic endothelial cells. Consequently, the p-Akt/Akt ratio decreased by 28% in the hypoxic endothelial cells cultured without HUCM in comparison with coronary endothelial cells cultured in normoxia (p < 0.05). In contrast, p-Akt and the ratio of p-Akt/Akt significantly increased by more than 60% in endothelial cells cultured with HUCM in comparison with endothelial cells cultured without HUCM in hypoxia (p < 0.05) (Fig. 2B). A representative Western blot of coronary artery endothelial cell p-Akt, Akt, and actin proteins during normoxia and hypoxia is shown in Figure 2A.

HUCM increases p-Akt in coronary endothelial cells and cardiac myocytes. Western blot expression of p-Akt, Akt, and actin in endothelial cells (A) and myocytes (C). Densitometry was used in the analyses of endothelial bands (B) and myocyte bands (D). The normoxic data are normalized to 1.0, and hypoxic data are expressed as a percentage of the normoxic data. NOR, normoxia; HY, hypoxia; HUCM, human umbilical cord blood cell hypoxic-conditioned media (*p < 0.05 or **p < 0.01 vs. NOR, #p < 0.05 vs. HY).
In cardiac myocytes cultured without HUCM during hypoxia, p-Akt decreased, and the p-Akt/Akt ratio declined by more than 55% in comparison with myocytes in normoxia (p < 0.005) (Fig. 2C, D). Akt and actin did not significantly change in these myocytes. The addition of HUCM to cardiac myocytes during 24 h of hypoxia prevented a significant decrease in p-Akt and a decrease in the p-Akt/Akt ratio during hypoxia. In cardiac myocytes treated with HUCM, the p-Akt/Akt ratio was more than 120% greater than the p-Akt/Akt ratio in myocytes cultured without HUCM (p < 0.02). The p-Akt/Akt ratio was not significantly different than the p-Akt/Akt ratio in myocytes in normoxia (Fig. 2D). A representative myocyte Western blot of p-Akt, Akt, and actin proteins during normoxia and hypoxia is shown in Figure 2C.
BCL-2 Is Increased in Endothelial Cells and Cardiac Myocytes Treated with HUCM
Figure 3A and B shows the antiapoptotic protein Bcl-2 in endothelial cells during normoxia and hypoxia. Phospho-Akt phosphorylates the apoptotic protein Bad in endothelial cells, which permits the dissociation of Bcl-2 from Bad and the Bcl-2′s antiapoptotic actions. In the present experiments, Bcl-2 decreased by 28% in endothelial cells cultured without HUCM during hypoxia in comparison with Bcl-2 in endothelial cells in normoxia (p < 0.05). In contrast, Bcl-2 expression increased by 34% in endothelial cells cultured with HUCM in which p-Akt was activated during hypoxia in comparison with endothelial cells cultured without HUCM in hypoxia (p < 0.05). In these experiments, the Bcl-2 in endothelial cells with HUCM during hypoxia was similar to the Bcl-2 in endothelial cells in normoxia.

HUCM increases Bcl-2 protein expression in coronary endothelial cells and cardiac myocytes. HUCM increases β-cell chronic lymphocytic leukemia (CLL)/lymphoma 2 (Bcl-2) protein expression in coronary endothelial cells (A, B) and cardiac myocytes (C, D) by Western blot analyses. Normoxic data are normalized to 1.0, and hypoxic data are expressed as a percentage of the normoxic data. NOR, normoxia; HY, hypoxia; HUCM, human umbilical cord blood cell hypoxic-conditioned media (*p < 0.05 or **p < 0.01 vs. NOR, #p < 0.05 or ##p < 0.01 vs. HY).
Figure 3C and D shows the antiapoptotic protein Bcl-2 in cardiac myocytes during normoxia and hypoxia. Hypoxia decreased Bcl-2 in cardiac myocytes by more than 35% (p < 0.01). In contrast, HUCM totally prevented the decrease of Bcl-2 in cardiac myocytes during hypoxia (p < 0.01). There was no significant difference between the Bcl-2 concentration in normoxic cardiac myocytes and the Bcl-2 concentration in cardiac myocytes cultured with HUCM during hypoxia.
HUCBC-Conditioned Media Inhibit Apoptosis
Hypoxic-induced apoptosis in coronary artery endothelial cells and cardiac myocytes was determined by Annexin V stains (Fig. 4). After 24 h of incubation in hypoxia, apoptotic FITC (green)-DAPI (blue) double-positive cells were counted with the use of fluorescence microscopy. Hypoxic treatment of endothelial cells cultured without HUCM increased endothelial cell apoptosis from 7.8% in normoxia to 13.8% in hypoxia (p < 0.002). However, HUCM, when added to endothelial cells during hypoxia, significantly decreased endothelial cell apoptosis from 13.8% to 9.5% (p < 0.05) (Fig. 4A, B). In these experiments, endothelial cell apoptosis with HUCM during hypoxia was not significantly different from endothelial cell apoptosis in normoxia.
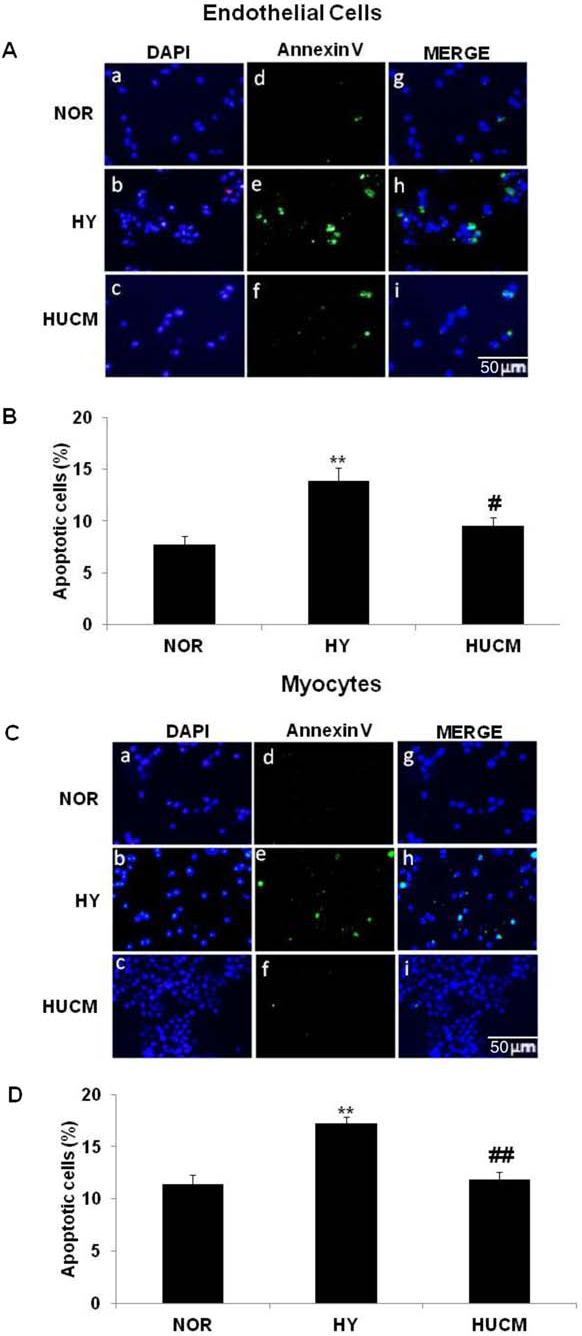
HUCM decreases apoptosis in coronary endothelial cells and cardiac myocytes as determined by Annexin V staining. Representative photomicrograph of endothelial cell nuclei stained with 4′,6′-diamidino-2-phenylindole (DAPI) in blue, endothelial cell apoptotic cell membranes stained green with Annexin V plus fluorescein isothiocyanate (FITC), and merged photographs during normoxia and hypoxia are shown in (A). Endothelial cells were visualized with magnification (200×). Scale bar: 50 μm. Quantification demonstrated that HUCM decreases endothelial cell apoptosis during hypoxia (B). Representative photomicrograph of myocyte nuclei stained with DAPI in blue, myocyte membranes stained with Annexin V plus FITC in green, and merged photographs (C). Myocytes were visualized with magnification (200×). Scale bar: 50 μm. Quantification revealed that HUCM protects myocytes against hypoxic-induced apoptosis (D). NOR, normoxia; HY, hypoxia; HUCM, human umbilical cord blood cell hypoxic-conditioned media. Mean ± SEM are presented (*p < 0.05 or **p < 0.01 vs. NOR, #p < 0.05 or ##p < 0.01 vs. HY).
Twenty-four hours of hypoxia also increased apoptosis in myocytes cultured without HUCM from 11.4% to 17.3% (p < 0.002). However, HUCM added to cardiac myocytes during hypoxia significantly prevented an increase in apoptosis. There was no significant difference in apoptosis between HUCM-treated myocytes during hypoxia and myocytes treated with normoxia (Fig. 4C, D).
To confirm that HUCM suppresses endothelial cell and cardiac myocyte apoptosis, we performed flow cytometry analyses for Annexin-positive cells. As shown in Figure 5A and B, endothelial apoptotic cells increased to 18.4% during 24 h of hypoxia compared with 13.9% during normoxia (p < 0.05). In contrast, HUCM significantly limited the increase in endothelial cell apoptosis during hypoxia such that there was no significant difference in the number of apoptotic cells in comparison with the normoxic endothelial cells.

HUCM decreases apoptosis in coronary endothelial cells and cardiac myocytes by Annexin V flow cytometry. Flow cytometry analysis of Annexin V staining (A) and quantification of the apoptotic endothelial cells (B). Flow cytometry analysis of Annexin V staining (C) and quantification of the apoptotic myocytes (D). HUCM decreased endothelial cell and myocyte apoptosis determined by flow cytometry. NOR, normoxia; HY, hypoxia; HUCM, human umbilical cord blood cell hypoxic-conditioned media. Mean ± SEM is presented (*p < 0.05 or **p < 0.01 vs. NOR, #p < 0.05 or ##p < 0.01 vs. HY).
In the flow cytometry analyses, hypoxia increased myocyte apoptosis to 27.9% compared to 10% apoptosis during normoxia incubation (p < 0.001) (Fig. 5C, D). However, HUCM significantly reduced the hypoxic-induced myocyte apoptosis from 27.9% in myocytes cultured without HUCM to 18.2% in myocytes cultures with HUCM (p < 0.05) (Fig. 5C, D).
AKT Inhibition Significantly Decreases HUCM Protection in Endothelial Cells and Cardiac Myocytes
We performed AKT inhibition studies with the sensitive and specific inhibitor API-1. API-1 inhibits Akt phosphorylation by binding to the PH domain of Akt and prevents phosphorylation at Thr 308 and Ser 473 (3,21,38). Consistent with our initial studies, 24 h of hypoxia significantly decreased Akt phosphorylation in coronary artery endothelial cells and cardiac myocytes in comparison with endothelial cells and myocytes in normoxia. This decrease in Akt phosphorylation in endothelial cells and myocytes was significantly prevented by HUCM. In contrast, incubation of endothelial cells or myocytes with HUCM plus API-1 significantly prevented the phosphorylation of Akt in endothelial cells (p < 0.04), as shown in Figure 6A and B, and in cardiac myocytes (p < 0.001), as shown in Figure 6C and D. Nonphosphorylated Akt expression remained unchanged in both endothelial cells and cardiac myocytes. We also determined caspase-3 activity in endothelial cells and cardiac myocytes because caspase is an important early indicator of apoptosis and has a critical role in stress-induced apoptosis. As shown in Figure 7A and B, caspase-3 activity increased by 29% in endothelial cells and by 30% in myocytes after 24 h of hypoxia (p < 0.05). In contrast, HUCM inhibited the increase in caspase-3 activity in endothelial cells and cardiac myocytes during hypoxia. However, the AKT inhibitor API-1 prevented the protective effect of HUCM in endothelial cells and cardiac myocytes such that the caspase-3 activity in these cells was nearly identical to the caspase-3 activity in endothelial cells and cardiac myocytes cultured without HUCM during hypoxia.

API-1 inhibits the expression of p-Akt in coronary artery endothelial cells and in cardiac myocytes. Endothelial cell Western blot bands (A) and myocyte bands (C) were analyzed by densitometer. Normoxic data in (B, D) are normalized to 1.0, and hypoxic data are expressed as a percentage of the normoxic data. NOR, normoxia; HY, hypoxia; HM, hypoxic-conditioned HUCBC media; HMI, hypoxic-conditioned HUCBC media + API-1. Mean ± SEM is presented (*p < 0.05 or **p < 0.01 vs. NOR, #p < 0.05 or ##p < 0.01 vs. HY, †p < 0.05 or ††p < 0.01 vs. HM).

The Akt inhibitor API-1 suppresses HUCM protection of endothelial cells and myocytes and increases caspase-3 expression. API-1 significantly increases endothelial cell (A) and myocyte apoptosis (B) in cells cultured with HUCM during hypoxia. Normoxic data are normalized to 1.0, and hypoxic data are expressed as a percentage of the normoxic data. NOR, normoxia; HY, hypoxia; HM, hypoxic-conditioned HUCBC media; HMI, hypoxic-conditioned HUCBC media + API-1. Mean ± SEM is presented (*p < 0.05 vs. NOR, #p < 0.05 vs. HY, †p < 0.05 vs. HM).
Discussion
The present experiments demonstrate that HUCBCs secrete growth factors and anti-inflammatory cytokines and that this HUCBC paracrine effect is significantly increased during hypoxia. Cord blood cell paracrine factors activate the cell survival protein Akt in coronary artery endothelial cells and cardiac myocytes during hypoxia. Activated Akt significantly increases the cellular antiapoptotic protein Bcl-2 and limits the cellular expression and activity of the apoptotic protein caspase-3 and the caspase cascade in coronary endothelial cells and cardiac myocytes. In this manner, HUCBCs can significantly decrease coronary artery endothelial cell and cardiac myocyte apoptosis.
HUCBC Growth Factors
Although HUCBCs have been reported to transdifferentiate into myocytes in ischemic muscles (33), HUCBC transdifferentiation to coronary endothelial cells and cardiac myocytes is not likely to occur acutely and does not readily explain the prompt beneficial effects of HUCBCs that we have observed in the treatment of acute myocardial infarctions and cardiomyopathy (12–17). In our studies of myocardial infarctions, HUCBCs significantly limited the myocardial expression of inflammatory cytokines by as much as 50% within 12 h after acute coronary artery occlusion and also significantly limited the infiltration into the left ventricle (LV) myocardium of inflammatory neutrophils and lymphocytes during the initial 12–72 h of acute myocardial infarction (17). These observations suggest that HUCBCs have significant paracrine actions whereby they release biologically active factors that limit the myocardial content of inflammatory cytokines and inflammatory cells and, in this manner, acutely limit the size of acute myocardial infarctions.
The present studies demonstrate that HUCBCs do secrete growth factors and anti-inflammatory cytokines, including angiopoietin, HGF, IL-4, IGF, PLGF, VEGF, angiogenin, SCF, IL-10, TIMP-1, and SDF that can have anti-inflammatory and cellular protective effects in acute myocardial infarctions. Moreover, the paracrine action of HUCBCs is significantly increased during severe hypoxia with 50–100% or more increases in the secretion of these factors. Additional HUCBC paracrine factors from proliferating HUCBCs in specialized cell culture media that are protective of cells include epidermal growth factor, platelet-derived growth factor, and fibroblastic growth factor (30). Since HUCBCs contain hematopoietic, endothelial, and mesenchymal stem cells, each of these stem cells can secrete growth factors and/or anti-inflammatory cytokines that can be beneficial to ischemic coronary artery endothelial cells and cardiac myocytes. Moreover, the effects of stem cell combinations or stem cells plus biomaterials or growth factors appear to be more beneficial than the effects of a single stem cell due to the facilitation of cell survival, growth, and proliferation during ischemia (4).
Growth factors, such as insulin-like growth factor, hepatocyte growth factor and vascular endothelial cell growth factor that are either overexpressed or injected in research animals during myocardial ischemia and infarction can significantly limit myocardial injury and LV remodeling by upregulating the expression and activation in myocytes and endothelial cells of the survival protein Akt (6,22,34,37). Akt is a cellular kinase that is important in cell growth, survival, and gene expression and has significant antiapoptotic activity (27,28,35). AKT activation reduces apoptotic cardiomyocyte death due to ischemia-reperfusion injury (2,35), pressure overload (5), and oxidative stress (1). However, inhibition or downregulation of Akt can prevent the protective effects of growth factors on cells and causes significant apoptosis and necrosis (35). Moreover, knockdown of Akt results in impairment in endothelial progenitor cell function and neovascularization in ischemic hind-limb muscles of research animals (27).
In the present experiments, HUCBC biologically active factors in conditioned media significantly activated Akt by more than 60% in coronary artery endothelial cells and cardiac myocytes and substantially increased cell survival during 24 h of severe hypoxia. In contrast, inhibition of Akt with API-1 significantly prevented the inhibitory effect of HUCBC media on endothelial cell and myocyte caspase activity and apoptosis. API-1 has been shown to be a highly sensitive and specific inhibitor of AKT phosphorylation and activation (3,21,38). In this regard, API-1 prevents Akt membrane localization and phosphorylation at Threonine 308 and Serine 473. In addition, API inhibits activation of Akt by growth factors such as insulin-like growth factor. However, API does not inhibit Ras-dependent extracellular signal-regulated kinase (ERK1/2), mitogen-activated protein kinase, c-Jun N-terminal kinases (JNK), protein kinase C, serine/threonine-protein kinase (SGK), or protein kinase A (3,21,38). The present experiments indicate that the biological factors released by HUCM activate Akt and that Akt activation is important in limiting hypoxic-induced endothelial cell and myocyte apoptosis.
Phospho-AKT
Phospho-Akt targets the downstream cellular apoptosis regulatory molecules proapoptotic Bad and the caspases (35,36). Phospho-Akt can phosphorylate Bad (28,35). The phosphorylated Bad then dissociates from the prosurvival Bcl-2, and Bcl-2 can limit or prevent the release of mitochondrial proteins into the cytoplasm of endothelial cells and myocytes (7,35). Treatment of cells previously transfected with Bad with a constitutively active Akt or activation of Akt by insulin-like growth factor can suppress Bad-mediated cell death by as much as 90% (7). Overexpression of Akt can also increase cardiomyocyte Bcl-2 expression and can protect myocyte mitochondrial membrane integrity by reducing free oxygen radical expression in hypoxic cells (28). Consequently, an important mechanism by which Akt inhibited endothelial cell and myocyte death in the present experiments is phosphorylation and inactivation of Bad and increased expression of the prosurvival protein Bcl-2.
Akt can also phosphorylate and activate in endothelial cells nitric oxide synthase (eNOS) and thereby increase nitric oxide (NO), which can upregulate antiapoptotic Bcl-2 in vascular endothelial cells and can nitrosylate caspases (9,11). In this manner, Akt-induced formation of NO can limit vascular endothelial cell and myocyte death.
Apoptosis in Acute Myocardial Infarction
Apoptosis, or programmed cell death, contributes to myocardial damage, most especially during the initial 6 h after coronary occlusion (20). In coronary artery endothelial cells and cardiac myocytes, hypoxic activation of the intrinsic death pathway causes Bad and Bax activation and (1) a fall in mitochondrial membrane potential and an increase in membrane permeability and (2) production of reactive oxygen species, activation of caspase and endonuclease that cause DNA fragmentation, increased intracellular calcium, cytoplasmic vacuolization, and increased cell membrane permeability (19). As a consequence, apoptotic bodies are formed. Activation of caspase-3 also leads to externalization of cellular phospholipid phosphatidylserine to the outer membrane of myocytes and endothelial cells within minutes after the onset of apoptosis. The amount of phosphatidylserine externalized on cells is proportional to the degree of caspase-3 activation (23).
Annexin V is a binding protein with high affinity for phosphatidylserine on myocyte and endothelial cell membranes and facilitates the diagnosis of apoptosis (23). Annexin V stains cells undergoing programmed cell death prior to morphological changes in the nucleus or fragmentation of DNA. Consequently, Annexin V can identify myocyte apoptosis after 30 min of myocardial ischemia (23). Moreover, Annexin determinations of apoptosis correlate well with other measures of apoptosis such as DNA flow cytometry and DNA electrophoresis (32). The present experiments demonstrate that HUCBCs can significantly inhibit coronary artery endothelial cell and myocyte apoptosis, when measured by Annexin cell staining and caspase-3 determinations, by secreting growth factors and anti-inflammatory cytokines that activate the cell survival proteins Akt and Bcl-2.
Footnotes
Acknowledgments
This work was supported in part by research facilities at the James A. Haley Medical Center and by grants from the Muscular Dystrophy Association and the Bugher Foundation. Human umbilical cord blood cells were provided by Saneron CCEL Therapeutics, Inc., Tampa, FL; CryoCell, Oldsmar, FL; and CordUse, Orlando, FL. P.R.S. is cofounder of Saneron CCEL Therapeutics, Inc. and is an inventor on cord blood-related patents licensed to Saneron. The contents of this publication do not necessarily represent the views of the Veterans Administration of the United States Government.