
Abstract
The expansion of pluripotent human embryonic stem cells (hESCs) requires a culture on feeder layers of mouse embryonic fibroblasts (MEFs). The culture model often causes immunogenic contaminations such as xenocarbohydrate, and inevitably forms teratoma in vivo. This study tested human umbilical cord-derived mesenchymal stem cells (HUCMSCs) as the feeder for hESCs. Wharton's jelly-derived HUCMSCs showed characteristics of MSCs and were easily maintained in a culture for over 20 passages. Under the mitomycin-inhibited HUCMSC feeder, hESCs maintained the features of embryonic stem cells (pluripotency and maintenance of normal karyotypes) after a prolonged culture of more than 20 passages. Notably, in extensive trials, no teratoma was formed in xenograft in NOD/SCID mice, but subsequent resumption of teratoma formation was noted upon transient coculturing with MEFs. Interestingly, among the four pluripotency-conferring genes, MYC and OCT4 were found to be downregulated in hESCs cocultured with HUCMSCs. Results of this study supported a nontumorigenic sustained culture of hESCs and did not form teratoma in vivo.
Introduction
Embryonic stem cells (ESCs) are derived from the inner cell mass (ICM) of the mammalian blastocyst and have a pluripotent developmental potential (13,24). In 1994, Bongso et al. described for the first time a culture of cells from the ICM of human blastocysts (3). The subsequent development of human embryonic stem cells (hESCs) (30,35) has since attracted much attention because of their potential application in regeneration medicine (27).
The sustained maintenance of hESCs depends on a coculture with a feeder layer of mouse embryonic fibroblasts (MEFs), which inevitably create the risk of transmitting nonhuman materials and unknown pathogens. Therapeutic products produced from such lines are therefore treated as xenografts and impose serious limitations on their clinical use. For example, hESCs that are grown on mouse feeder cells and in animal-derived cell culture medium are reportedly contaminated with xenocarbohydrate N-glycolyneuraminic acid (Neu5Gc) (25). These and other immunogenic contaminations are difficult to eliminate from human cell lines that are cocultured with animal cells. To solve this problem, hESCs have been successfully cocultured with human fetal muscle and skin cells (31), adult fallopian tube epithelial cells (31), foreskin cells (1,19), and bone marrow stem cells (8).
Teratoma formation is another frequently raised concern associated with hESC-derived cell therapies. When hESCs are injected into an ectopic site in immunocompromised mice, they form a teratoma that comprises differentiated tissues originated from all three embryonic germ layers. Although the possibility of tumorigenesis is much lower when differentiated hESCs are used therapeutically, contamination with residual undifferentiated hESCs is inevitable and difficult to detect. Teratomas have been observed in various mouse and primate ESCs-derived differentiated cell preparations transplanted in animal models (6,15,33).
Wharton's jelly (WJ) of the umbilical cord exhibits the characteristics of stromal cells and is a novel source of mesenchymal stem cells (26). Mesenchymal stem cells that are derived from WJ of human umbilical cord (HUCMSCs) have been shown to differentiate into neural-like cells, osteocytes, and adipocytes both in vitro and in vivo (12,26,37). In this study, the efficacy of HUCMSCs as feeders in hESCs culture is tested and the nontumorigenic feature of this coculture system is demonstrated.
Materials and Methods
Isolation and Expansion of HUCMSCs
Human umbilical cord samples (20 cm in length, 20 g in weight) were collected in sterile boxes that contained Hanks' balanced salt solution (HBSS; Gibco/BRL 14185-052). The protocols for collecting and using human umbilical cord were approved by the Institutional Review Board of Buddhist Tzu Chi General Hospital. Written informed consent was obtained from the pregnant women before labor.
Collected human umbilical cord tissues were washed three times in Ca2+ and Mg2+-free PBS (DPBS, Life Technology). They were mechanically cut along the midline, and the umbilical arteries, vein, and outlining amniotic membrane were dissociated from the WJ. The jelly was then cut into pieces that were smaller than 0.5 cm3, treated with trypsin/EDTA (Sigma, St. Louis, MO), and incubated for 30 min at 37°C in a 95% air/5% CO2 humidified atmosphere. The explants were then cultured in Dulbecco's modified Eagles medium (DMEM) that contained 10% human umbilical cord blood serum (CBS) and antibiotics and left undisturbed for 5–7 days to allow the cells to migrate from the explants.
The MSC-specific surface markers were characterized by flow cytometric analysis. The cells were detached using 2 mM EDTA in PBS, washed, and incubated with the appropriate antibody that was conjugated with fluorescein isothiocyanate (FITC) or phycoerythrin (PE). The antibodies were cluster of differentiation 1q (CD1q), CD3, CD10, CD13, CD14, CD31, CD34, CD45, CD90, CD73, CD56, human leukocyte antigen (HLA)-ABC, HLA-DR, CD49b, CD49d, CD29, CD44, CD105, CD117, and CD166 (BD, PharMingen). Then the cells were analyzed using a Becton Dickinson flow cytometer (Vantage SE, Becton Dickinson, San Jose, CA).
In Vitro Differentiation Assay for HUCMSCs
To induce osteogenic and adipogenic differentiations, HUCMSCs were transferred to an osteogenic medium (DMEM supplemented with 10% CBS, 0.1 μmol/L dexamethasone, 10 mmol/L β-glycerol phosphate, 50 μmol/L ascorbate), and adipogenic medium (DMEM supplemented with 10% CBS, 1 μmol/L dexamethasone, 5 μg/ mL insulin, 0.5 mmol/L isobutylmethylxanthine, and 60 μmol/L indomethacin) for 3 weeks. The potential for osteogenesis was evaluated by determining the mineralization of calcium by staining with Alizarin Red S (Sigma). To assess adipogenic differentiation, intracellular lipid droplets were observed under a microscope and the lipid droplet was verified by staining with Oil Red O (23).
Culture of Human ES Cells
The hESCs (TW1 cell line; p22) (9) were obtained from Biomedical Technology and Device Research Laboratories, Industrial Technology Research Institute and initially cultured on MEFs, as directed by the supplier (FIRDI, Taiwan, http://www.firdi.org.tw/). Either MEFs (p3) or HUCMSCs after mitomycin-C deactivation were used as feeder cells for culturing the hESCs. They were plated at a density of 200,000 cells per 9.4-cm2 well in six-well plates. The hESC culture medium comprised 80% (v/v) knockout (KO) DMEM, 20% (v/v) KO serum replacement, 2 mM l-glutamine, 10 mM nonessential amino acids (all from Invitrogen), 50 μM β-mercaptoethanol (Sigma), and 4 ng/ml basic fibroblast growth factor (bFGF).
Characterization of hESCs
The hESCs were characterized by immunocytochemistry using fluorescence-labeled antibodies specific for undifferentiated hESCs, which were stage-specific embryonic antigen-1 (SSEA-4), SSEA-1, TRA-1-60, TRA-1-81, and octamer-binding transcription factor 4 (Oct-4) (ES Cell Characterization Kit; Chemicon). Initially, hESCs were cultured on chamber slides (Nunc, Denmark) or sterile cover glasses (Assistent, Germany) in culture dishes with feeder cells. At specified times on days 3–7 following passage, the colonies of the hESCs were subjected to immunofluorescence staining. Briefly, cells were fixed with 4% paraformaldehyde and then underwent several procedures, including permeabilization (0.1% Triton X-100), blocking (4% normal goat serum), treatment with primary antibody (1:10–1:50 dilution), three washings, treatment with fluorescent-labeled secondary antibody, three more washings, covering with a coverslip, and mounting. Alkaline phosphatase (AP) staining was conducted using the ES Cell Characterization Kit (Chemicon).
Differentiation of hESCs
Before differentiation, hESCs were collected and resuspended in the hESC medium in the absence of bFGF. The hESCs were then cultured in low attachment six-well plates as aggregates in suspension. After 5 days, fetal bovine serum (FBS; 5% final) was added. Aggregated hESCs usually formed an embryoid body (EB) after 7–10 days, and mature (cystic) EBs subsequently emerged from 20–80% of the formed EBs. The resultant solid or cystic EB was then plated on gelatin-treated chamber slides or 35-mm culture dishes for further differentiation and was then subsequently treated similarly to those cells that were grown directly in an adherent culture. After fixation, the differentiated hESCs were studied by immunocytochemistry using fluorescence-labeled antibodies specific for three embryonic germ layers, which were ectoderm [microtubule associated protein 2 (MAP2), neurofilament 200 (NF200; Chemicon)], mesoderm (brachyury), and endoderm [AT motif-binding factor 1 (ATBF1; Santa Cruz)].
Karyotyping of hESCs
The karyotypes of cells were studied at passage 10. On day 7 after passage, hESCs were treated with 0.1 μg/ml colcemid (Gibco) for 4 h. After the cells were washed, they were treated with either 0.25% trypsin for 3–5 min or collagenase type IV for 8 min, pipette, and harvested. They were then fixed and mounted on glass slides. The metaphases were analyzed using the standard G-banding method in a certified cytogenetic laboratory.
Teratoma Formation
hESCs were detached with mechanical slicing using glass capillaries, pelleted, resuspended in PBS with Matrigel (BD Biosciences) (1:1), and injected into the back subcutaneous tissue (n = 17) or renal capsule (n = 4) of nonobese diabetic-severe combined immunodeficient (NOD-SCID) mice. Cells were counted using a hemocytometer and suspended in PBS with Matrigel (1:1) in various concentrations (18,22,28). hESCs were kept on ice <45 min for optimal viability prior to injection. The teratoma formation was followed up by palpation and resulting tumors were dissected, fixed, embedded in paraffin, and processed for histology.
RT-PCR and Quantitative RT-PCR
Undifferentiated or differentiated hESCs that had been cultured on HUCMSCs or MEF feeder layers were removed mechanically and treated with RLT lysis buffer (Qiagen). The first strand of cDNA was synthesized using a SuperScript III One-Step RT-PCR kit (Invitrogen) following the manufacturer's instructions. Table 1 presents the sequence, annealing temperature, and product size of each pair of primers. All PCR samples were analyzed by electrophoresis on 2% agarose gel that contained 0.5 μg/ml ethidium bromide (Sigma). For quantitative RT-PCR (qRT-PCR) analysis, FastStart universal SYBR green master (ROX, Roche, USA) gene expression assays was used in an ABI Step One Plus system (Applied Biosystems), with GAPDH used as an internal control. The sequences of primers and annealing temperatures are showed in Table 1.
PCR Primers Used to Characterize Human Embryonic Stem Cells (hESCs) Before and After Differentiation

Western Blot Assay
Cells were lysed in the lysis buffer [150 mM NaCl, 50 mM Tris-HCl (pH 7.4)], 1% Nonidet P-40] plus proteinase inhibitor cocktail (Roche, Indianapolis, IN). Proteins were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane (Hybond-C Super; Amersham, Little Chalfont, UK). The membranes were incubated with anti-c-myc (2 μg/m) or anti-α-actin (1:10,000; Sigma-Aldrich) monoclonal antibodies. Horse radish peroxidase (HRP)-conjugated goat anti-mouse IgG (Jackson Immuno-Research Laboratories) was used as the secondary antibody. Bound antibodies were detected using enhanced chemiluminescence reagents (ECL; Amersham).
Results
Characteristics of HUCMSCs Derived from Human Umbilical Cord
Upon the first culturing of WJ tissue pieces, attached growing cells with a spindle-shaped morphology migrated from the explants (Fig. 1A). These cells divided rapidly with a doubling time of 28 h, and underwent more than 25 passages, equivalent to over 40 population doublings, without spontaneous differentiation (data not shown). They were negative for CD1q, CD3, CD34, CD45, CD56, CD117, and HLA-DR, and positive for CD13, CD29, CD44, CD73, CD90, CD166, and HLA-ABC (Fig. 1B). These observations demonstrate that cells that are isolated from WJ of the human umbilical cord have the same surface markers as MSCs (29).

Morphology, immunotyping, and in vitro differentiation of human umbilical cord-derived mesenchymal stem cells (HUCMSCs). Cells of Wharton's jelly growing from explants are fibroblast-like with a spindle-shaped morphology (A). Flow cytometry of rapidly dividing HUCMSCs showed negative for cluster of differentiation 1q (CD1q), CD3, CD34, CD45, CD56, CD117, and human leukocyte antigen (HLA)-DR, and positive for CD10, CD13, CD29, CD44, CD73, CD90, CD166, and HLA-ABC (B). Upon adipogenic differentiation, the cells formed neutral lipid vacuoles and contained numerous Oil Red O-positive lipid droplets (C, upper panel). In osteogenic medium, the cells broadened to form a mineralized matrix, which was strongly stained with Alizarin Red S (C, lower panel) after 3–4 weeks of cultivation. Expression of genes specific for adipogenic (peroxisome proliferator-activated receptor; PPARγ) and osteogenic (osteopontin) differentiation was showed by RT-PCR analysis with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a positive control (D). Scale bar: 1000 μm (A, left panel), 100 μm (A, right two panels and C).
In Vitro Differentiation of HUCMSCs to Osteocytes and Adipocytes
Adipogenic differentiation of HUCMSCs was apparent 2 weeks after incubation with an adipogenic medium supplement. At the end of the second week, changes were evident in the cell morphology, and in the formation of neutral lipid vacuoles: almost all cells contained many Oil Red O-positive lipid droplets (Fig. 1C). Similarly, the induction of differentiation in the osteogenic medium caused the treated cells to grow rapidly and contain mineralized matrices, which were strongly stained by Alizarin Red S, indicating deposition of calcium after 3–4 weeks of cultivation (Fig. 1C). Expression of adipogenic (peroxisome proliferative activated receptor; PPARγ) and osteogenic (osteopontin) genes was evident in RT-PCR analysis (Fig. 1D).
Characterization of HUCMSC Cocultured hESCs
The hESCs that were transferred to the HUCMSC feeders formed colonies effectively and continued to proliferate with a doubling time of 36 h. The morphology of colonies differed slightly from that of those that were cultured on MEFs (Fig. 2A, B). However, the individual hESC morphology cultured on HUCMSCs remained the same as on MEFs. The cells remained round and small, with a high nucleus/cytoplasm ratio, and the notable presence of one to three nucleoli (Fig. 2C, D). The TW1 hESCs on HUCMSC feeders expressed AP, Oct-4, SSEA-4, TRA-1-60, and TRA-1-81 markers (Fig. 3A–E). At passage 20, they revealed normal karyotypes of 46, XX (Fig. 3F).

Morphology of undifferentiated human embryonic stem cell (hESC) colonies grown on HUCMSC and mouse embryonic fibroblast (MEF) feeders. Pictures of hESC colony grown on HUCMSCs (A) and MEF (B) feeder layers are showed. A magnification of colony grown on HUCMSCs revealed typical hESC morphology with high nucleus/cytoplasm ratio (C, D). Scale bar: 1000 μm (A, B), 100 μm (C, D).
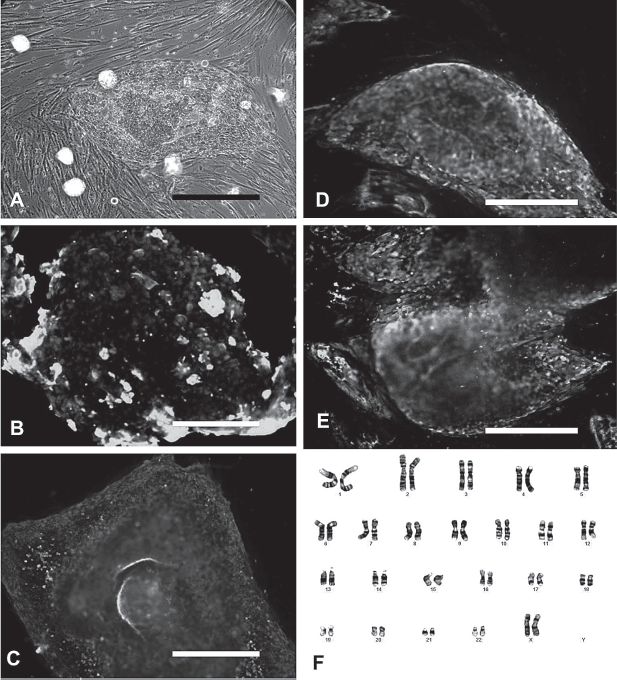
Phenotypes of human embryonic stem cells (hESCs) cultured on HUCMSCs. Immunostaining of hESC colony with specific antibodies revealed strong expression of alkaline phosphatase (A), octamer binding transcription factor 4 (Oct4) (B), stage-specific embryonic antigen (SSEA)-4 (C), TRA-1-60 (D), and TRA-1-81 (E). A representative normal karyotype (46, XX) was observed in hESCs after 20 passages of continuous culture on HUCMSCs (F). Scale bar: 1000 μm.
In Vitro Pluripotency of HUCMSC Cocultured hESC Line
Like those grown on MEFs, hESCs cultured with HUCMSC feeders formed EBs when cultured in suspension. Within these EBs, cell differentiation that represented three embryonic germ layers was observed. These cells expressed ectodermal markers (MAP-2 and NF-200), mesodermal marker (brachyury), and endodermal marker (ATBF1), as indicated by immunohistochemistry (Fig. 4). Cells that were harvested from 7 to 10-day-old EBs expressed such genes as βIII tubulin (ectoderm), heart- and neural crest derivatives-expressed protein 1 (Hand1; mesoderm), GATA4 (endoderm), and growth differentiation factor 9 (GDF9; germ cell related), as indicated by RT-PCR (Fig. 5).
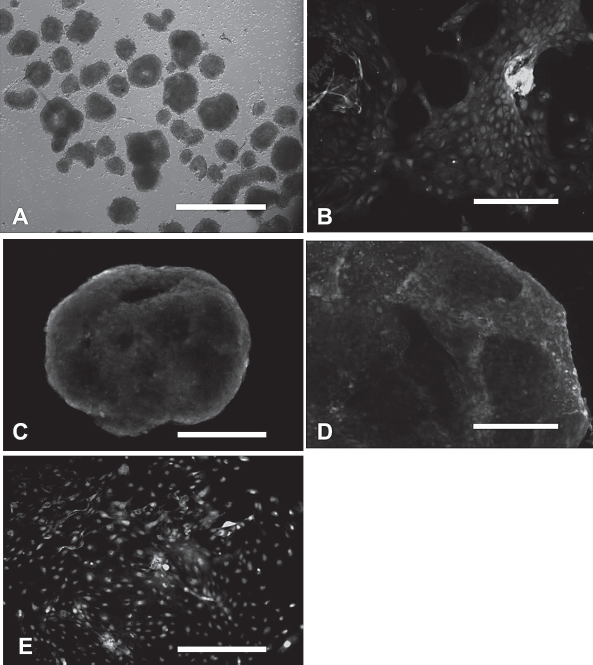
Fluorescence immunostaining of embryonic bodies (EBs) derived from human embryonic stem cells (hESCs). Embryonic bodies are showed under phase contrast microscope (A), and by immunostaining with antibodies against neurofilament (NF)-200 (B), brachyury (C), AT motif binding factor 1 (ATBF1) (D), and microtubule associated protein 2 (MAP2) (E). Scale bar: 1000 μm.

Expression of differentiation markers in human embryonic stem cells (hESCs) on different feeders. (A) Expression of genes that are specific for germ cell (growth differentiation factor 9; GDF9), ectoderm (βIII tubulin), mesoderm (heart- and neural crest derivatives-expressed protein 1; HAND1), and endoderm (GATA4) was showed by RT-PCR. GAPDH was used as a control. (B) Semiquantitative analysis of gene expression in (A). EB, embryoid body; MEF, mouse embryonic fibroblast; WJ, Wharton jelly (UCMSC).
Nontumorigenesis and In Vivo Pluripotency of HUCMSC Cocultured hESCs
The developmental potential of hESCs after long-term culture on HUCMSC feeders was investigated in vivo using a xenograft model. In extensive trials with 21 different transplantations in both NOD/SCID (n = 18) and nude (n = 3) mice, no teratoma growth was observed during long-term follow-up for over 3 months (Tables 2 and 3). However, teratomas were clearly observed upon transient (days) subsequent culturing on MEF feeders. The histologies of the teratomas that were derived from the hESCs before and after they were transferred to the HUCMSC feeder did not differ (Table 2, Fig. 6).

Teratomas developed from human embryonic stem cells (hESCs) with a change of the feeder cells from HUCMSCs to MEFs. Teratoma (arrow) readily developed from hESCs after change of the feeder from HUCMSCs to MEFs on nonobese diabetic severely combined immunodeficient (NOD-SCID) mice. Histological section revealed ribbon of melanocytes with a retina-like structure (B), neurotube-like structures (C–E), odontogenic epithelium (F), neuroepithelium (G), immature cartilage (H), and immature squamoid epithelium (I). Scale bar: 100 μm.
Summary of Xenograft Experiments With TW1 Human Embryonic Stem Cells Alternatively Cultured on MEFs and HUCMSCs as Feeders

HUCMSCs, human umbilical cord mesenchymal stem cells; MEFs, mouse embryonic fibroblasts.
Trials of Xenograft Tumorigenesis of Human Embryonic Stem Cells Cocultured With HUCMSCs
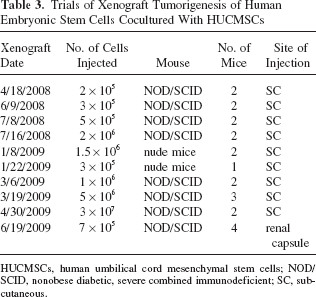
HUCMSCs, human umbilical cord mesenchymal stem cells; NOD/SCID, nonobese diabetic, severe combined immunodeficient; SC, subcutaneous.
Downregulation of MYC in hESCs Cocultured with HUCMSCs
We further tested the expression of key pluripotency genes (32) in hESCs on the two different feeders. Markers of undifferentiated stem cells, such as Oct-4, Nanog, and sex determining region Y)-box 2 (Sox2), were readily expressed as indicated by RT-PCR (Fig. 7A, B). Lower expression of the homeobox gene OCT4 and proto-oncogene MYC was observed in hESCs cocultured with HUCMSCs (Fig. 7A, B). This downregulation of MYC was further demonstrated by Western blot analyses and quantitative RT-PCR (Fig. 7C, D).

Expression of pluripotent genes in human embryonic stem cells (hESCs) cultured on HUCMSCs and MEFs. (A) RT-PCR analysis of OCT4, NANOG, sex-determined region box 2 (SOX2), and MYC in hESCs (ES) cultured on HUCMSCs (WJ) and MEFs with GAPDH as the internal control. (B) Semiquantitative analysis of gene expression in (A). (C) C-myc protein was measured by Western blot analysis. (D) The mRNA levels of MYC were measured by qRT-PCR as revealed by folds of the internal control. EB, embryoid body.
Discussion
The culture system that is described herein this work provides completely animal-free conditions for the growth of hESCs. HUCMSCs used as feeders supported the sustained undifferentiated growth of the TW1 human ESC line for more than 90 passages. The characteristics of hESCs were maintained. The lines that were cultured with HUCMSC feeders were strongly positive for surface markers that are also characteristic of undifferentiated primate ESCs (SSEA4, TRA-1-60, and TRA-1-81), but were negative for SSEA1. When they were removed from the HUCMSC feeder and grown in suspension, they spontaneously differentiated into various cell types, including representative types from the three embryonic germ layers. In earlier works, differentiated or nondifferentiated human feeders of both fetal and adult origins, including human fetal muscle, fetal skin, adult fibroblast, and bone marrow stem cells, have been successfully used to maintain hESCs. However, all of them give rise to teratoma in xenograft (1,8,19,31).
A striking finding of this work is the nontumorigenic feature of hESCs cultured under a HUCMSC feeder. No teratoma growth was observed in extensive trials with various strains of immune-compromised mice, various injection doses, as well as coinjection with Matrigel, which was showed to enhance teratoma growth (28). Most importantly, the stem cells readily resume their teratoma-forming activity during a short period of culturing on the MEF feeder.
Previously, human umbilical cord stromal cells (10) and amnion epithelial cells (21) have been used to support self-renewal growth of hESCs, and both grew teratomas in immune compromised mice. Table 4 compares protocols used for cultivate feeder cells and for coculture with hESCs in these two studies and ours. As seen, slight differences of culture ingredients in the preparation of feeder and coculture may result differently with regards to teratoma formation.
Comparison of the Coculture Conditions With Two Other Fetal Origins of Feeders

CBS, umbilical cord blood serum; bFGF, basic fibroblast growth factor; FBS, fetal bovine serum; hLIF, human leukemia inhibitory factor.
Teratoma formation has long been regarded as an in vivo proof of pluripotency of stem cells derived from sources varying from inner cell mass, epiblast, embryonic germ cells, and adult germline cells (5). This study, for the first time, demonstrated that pluripotency may not be teratoma forming. In the development of the human embryo, ESCs do not form tumors. The microenvironment provided by HUCMSCs in the reported coculture state may more resemble that of real life.
HUCMSC supported hESCs seemed to be less efficient at differentiating to germ cell and mesoderm lineages, but had a comparable efficiency in differentiating to ectoderm and endoderm (Fig. 5B). This heterogeneous expression of early lineage-commitment markers is in reminiscence of murine epiblast-derived stem cells (EpiSCs) (34). The epiblast origin of HUCMSCs is assumed to be unique. Epiblast is the primary derivative of the inner cell mass and develops to the three germ layers that form the embryo. Umbilical cord is derived from the very early mesoderm of epiblast. Recently, EpiSCs from mouse and rat were found to resemble hESCs in terms of morphology, growth factor requirement, and gene expression patterns (20). Mouse ESCs and EpiSCs are interconvertible through changes of culture conditions (11,17), and this also happens in human pluripotent cells (4,16,38). We speculate that, differing from other feeders that are derived from differentiated germ layers, HUCMSCs, when cultured in specific a condition (Table 4), may support a transition of hESCs to EpiSCs, which does not grow teratoma in vivo. This transiently differentiated state of EpiSCs is readily converted back to the ESC state upon the change of feeder back to MEFs.
The use of human umbilical cord as the source of feeder cells has several advantages. Human umbilical cord is widely available, easy to handle, and of low immunogenicity (2,12,14). As revealed in this work, HUCMSCs are highly proliferative and can undergo more than 20 passages without senescence. MSCs derived from Walton's jelly of umbilical cord are a highly promising source for cellular therapy and can be differentiated into islet cells, dopamine neurons, peripheral neurons, and liver cells (7,36,39).
In summary, this work demonstrated a safe, feasible, and robust coculture system for the sustained culture of undifferentiated hESCs. The proposed system significantly reduces the workload that is involved in the preparation of new feeder lines. Most importantly, we have described, for the first time, a set of culture conditions for hESCs that appears to eliminate the most feared characteristic of transplantation of hESCs—their ability to form tumors. This feeder system may therefore support significant advancement in the future applications in cell therapy.
Conclusion
We have described, for the first time, a set of culture conditions for hESCs that appears to eliminate their most feared characteristic when it comes to transplantation—their ability to form tumors. Besides, HUCMSCs can be easily derived and maintained. Under the above culture conditions, it may support a sustained culture of hESCs that does not form teratoma in vivo.
Footnotes
Acknowledgments
The authors would like to thank the National Science Council of the Republic of China, Taiwan, for financially supporting this research under the intramural project of Buddhist Tzu Chi General Hospital (TCRD 98-36). The authors also thank Ted Knoy for his editorial assistance. The authors declare no conflicts of interest.