
Abstract
Recently, FDA approved the clinical use of autologous fibroblasts (LAVIV™) for the improvement of nasolabial fold wrinkles in adults. The use of autologous fibroblasts for the augmentation of dermal and subcutaneous defects represents a potentially exciting natural alternative to the use of other filler materials for its long-term corrective ability and absence of allergic adverse effects proved by clinical application. However, compared to the clinical evidence, preclinical studies are far from enough. In this study, human skin-derived fibroblasts were cultured and expanded for both in vitro and in vivo observations. In vitro, the subcultured fibroblasts were divided into two groups. One set of cells underwent cell cycle and karyotype analysis at passages 5 and 10. The second group of cells was cocultured in medium with different concentrations of human skin extract D for the measurement of collagen concentration and cell count. In vivo, the subcultured fibroblasts were injected into nude mice subcutaneously. Biopsies were taken for morphology observation and specific collagen staining at 1, 2, and 3 months after injection. The results in vitro showed no significant differences in cell cycle distribution between passages 5 and 10. Cell proliferation and secretion were inhibited as the concentration of extract D increased. In vivo, the fibroblasts were remarkably denser on the experimental side with no dysplastic cells. Mitotic cells were easily observed at the end of the first month but were rare at the end of the third month. Type III collagen was detected at the end of the first month, while collagen type I was positive at the end of the second month. The content of both collagens increased as time passed. The above results indicated that the use of the autologous fibroblasts was safe, providing a basic support for clinical use of fibroblasts.
Introduction
The development and use of materials for correction of dermal defects have exploded over the past several decades, along with patient demand and manufacturer interest. Since bovine collagen was used as the first injectable material to treat rhytides and scars in 1977 (9), more and more implant substances, either biological or synthetic, have been made available for treating dermal and subcutaneous defects. However, unwanted side effects such as swelling, bruising, erythema, hypersensitivity, and infection were observed with all products used, even in the most experienced practitioners' hands. Moreover, virtually all biological materials are ultimately resorbed, and synthetic materials may lead to unpleasant complications, including migration, granuloma formation, late allergic reactions, and rough surface (13). Therefore, they might not be the most ideal material for permanent soft tissue augmentation, especially on the face (6).
Clinical use of injectable autologous skin-derived fibroblasts was first started by Isolagen Technologies in 1995 to repair dermal and subcutaneous contour deformity. Longterm correction and the absence of allergic adverse effects have been reported (1,2,12,17), which made autologous fibroblasts a promising alternative to the use of processed bovine collagen and other foreign materials. A prospective, placebo-controlled, phase III clinical trial has also shown that autologous fibroblast injections can safely and effectively produce improvements in rhytides, acne scars, and other dermal defects that continue for at least 12 months after injection (18).
However, despite the clinical evidence, the preclinical studies of autologous human skin-derived fibroblasts are insufficient. Although the FDA finally approved the clinical use of autologous cultured fibroblasts (LAVIV™; Fibrocell Science, Inc., Exton, PA, USA) for the improvement of the appearance of moderate to severe nasolabial fold wrinkles in adults on June 21, 2011 (16), based on little preclinical data (8,20), the Committee also commented on the lack of sufficient data related to the processing, characterization, and collagen production of the injected cells and insufficient information to assess the safety of the product (16).
In the past 10 years, we have been dedicated to proving the safety and efficacy of autologous fibroblasts. In our previous research (20), by using a tritiated thymidine (3H-TdR) labeling technique, we have demonstrated that the implanted cultured autologous fibroblasts could survive in vivo for more than 5 months and actively secrete new collagen.
This study is a continuous work of the previous one. In this study, our concern is to provide evidence of the preclinical safety of using human skin-derived fibroblasts and to demonstrate their proliferation and secretion activity. Both in vitro and in vivo studies were performed with the following objectives: (1) to prove the genomic stability of in vitro cultured human skin-derived fibroblasts by cell cycle analysis and karyotype analysis; (2) to conform that the proliferation and secretion of the cells are regulated after transplantation by in vitro cocultured with skin extract D and in vivo observation; and (3) to observe in vivo growth characteristics and collagen secretion activity of the cultured human skin fibroblasts after transplanted into nude mouse.
Materials and Methods
Study Design
This study was approved by the Chinese People's Liberation Army General Hospital's Ethics Committee. It consisted of both in vitro and in vivo parts (Fig. 1). The in vitro side looked at fibroblasts derived from human skin that were cultured for genomic stability and growth regulation tests. The in vivo aspect explored cultured human skin fibroblasts after subcutaneous injection into nude mice to investigate the growth and collagen secretion ability.
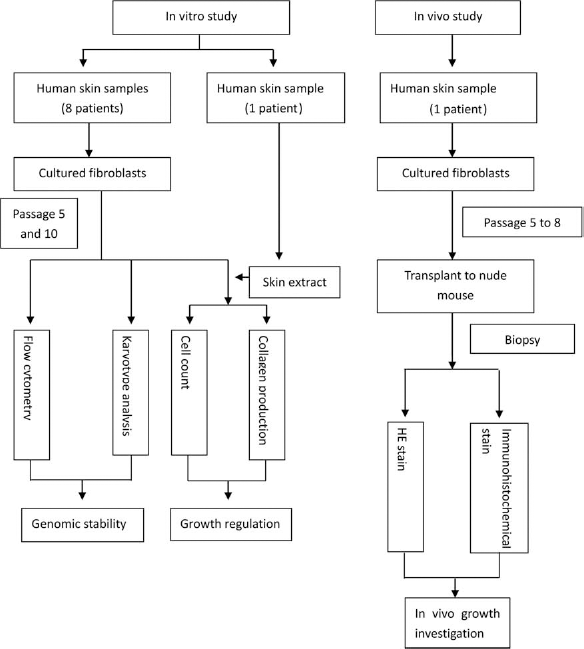
Flow chart of study design: Preclinical safety studies on autologous cultured human skin fibroblast transplantation. H&E, hematoxylin and eosin.
Samples and Animals
Human skin samples used for generating cell lines for in vitro studies were obtained from eight patients (three men and five women; mean age, 28.5 years; range, 18 to 45 years) undergoing blepharoplasty in our department. The human skin sample for in vivo was obtained from a 28-year-old female during upper blepharoplasty. The human skin sample for preparing skin extracts was obtained from a 32-year-old female during abdominoplasty. All volunteers had signed consent forms preoperatively and were evaluated to rule out histories of heavy smoking, alcohol abuse, drug abuse, diabetes mellitus, autoimmune diseases, or steroid treatment.
Six 2-week-old BALB/CNU nude mice (three males and three females) were obtained from the Experimental Animal Center of the Peking Union Medical College (Beijing, China). Animal weights at the initiation of study ranged from 22 to 29 g. All animal experiments were performed in compliance with the college research council's guidelines for animal care and were approved by the Animal Experiments Committee of Peking Union Medical College.
Cell Culture
Preparation of the specimens and primary cultivation of autologous skin fibroblasts were described in our previous article (20). When primary cultured fibroblasts reached confluence, the adherent cells were treated with 0.25% trypsin (HyClone, Logan, UT, USA) at 37°C for 2–3 min. The detached fibroblasts were centrifuged (230 × g for 8 min) and resuspended in Dulbecco's modified Eagle's medium (DMEM; Gibco, Gaithersburg, MD, USA) containing 10% fetal bovine serum (FBS, HyClone). The cells were then subcultured in 175-cm2 flasks (Nunc, Penfield, NY, USA) and passaged every 5 days at a split ratio of 1:3.
Genomic Stability of In Vitro Cultured Human Fibroblasts (Performed on Cells at Both Passages 5 and 10)
Cell Cycle Analysis
Subcultured cells were digested, centrifuged (230 × g for 8 min), and resuspended in phosphate-buffered saline (PBS, HyClone) when they reached 95% confluence. Ethanol (95%) was added to the suspension to permeabilize the cells. The suspension was stored at 4°C for at least 24 h. Two to three hours before analysis, the cells were centrifuged (230 × g for 5 min), washed three times with PBS, and centrifuged again. The supernatant was aspirated, and 1 ml propidium iodide (PI, Sigma-Aldrich, St. Louis, MO, USA) staining solution (50 μg/ml) was added in for PI staining of the DNA to determine cell cycle. The sample was stored at 4°C for 40 min and then analyzed using a Beckman Coulter EPICS XL Flow Cytometer (Beckman Coulter, Brea, CA, USA). Differences between the two passages were analyzed by Wilcoxon matched-pairs signed-ranks test using SPSS 13.0 (IBM, Armonk, NY, USA). Statistical significance was taken when values were p < 0.05.
Karyotype Analysis
Seventy-two hours after passage, cells were blocked in the metaphase stage of mitosis by addition of Colcemid (Sigma-Aldrich) to a final concentration of 0.4 μg/ml and incubated at 37°C for 6 h. The cells were then digested and resuspended with 0.075 M KCl (Yili Chemicals, Beijing, China). After being cultured at 37°C for 15 min, the cells were centrifuged (230 × g for 8 min) and resuspended again in 0.5 ml 0.075 M KCl. One milliliter of freshly prepared fixative [absolute methanol and glacial acetic acid (both Yili Chemicals); 3:1 mixture] was then added and mixed gently. The suspension was then left for 30 min at room temperature for fixation before centrifuged again at 230 × g for 2 min. The supernatant was removed, and the cells were resuspended in fresh fixative. The fixed cells are dropped on a precooled 4°C microscope slide (Feizhou Glass, Jiangsu, China), dried, and stained with acetoorcein (Yili Chemicals). Appropriate chromosome spreads were photographed, cut out, and arranged according to size and position of the centromere to observe their general morphology. In each samples, 50 cells in metaphase were randomly chosen for observation.
Growth Regulation Test at the Presence of Skin Extracts (Performed on Cells at Passage 5)
Preparation of Skin Extracts
Skin extracts were made as previously described (11) with little modifications. The skin sample used for preparing skin extracts was trimmed by removing the subcutaneous adipose tissue first and then cutting into small pieces, about 5 × 5 mm. These small pieces of skin were frozen in liquid nitrogen and pulverized in a cooled Culatti mill (IKA, Guangzhou, China). The resulting powder was extracted by serial extraction at a ratio of 2.5% (w/v) with doubly pure water, NaCl (0.15 M), NaCl (1 M), citric acid/citrate buffer (1 M, pH 3.5; all Yili Chemicals), and urea (6 M; Sigma-Aldrich) sequentially. The supernatant was removed and transferred into a dialysis bag (Huichun Medical Equipment, Jiangsu, China) and dialyzed for 48 h in 2 L of ultrapure water (Merck, Beijing, China), which was changed three times during the 48-h period. The resulting solution was then lyophilized to obtain extract D as a pale powder. The extracts were stored at −20°C until required.
Preparation of Media Containing Different Concentrations of Extract D
Different quantities of stored extract were dissolved in DMEM supplemented with 10% FBS to create a serial media with different concentrations of extract D at 2, 1, 0.5, 0.25, and 0.125 mg/ml, respectively. The medium was passed through a 0.2-μm filter (Corning, Corning, NY, USA) and kept in 4°C.
Cell Culture with Extract D
Subcultured cells at passage 5 were trypsinized and centrifuged (230 × g for 8 min), and the cell pellet was suspended in 500 μl of DMEM supplemented with 10% FBS. A 10-μl suspension of cells diluted with 0.05% Evan's blue dye (Sigma-Aldrich) in a 1:9 ratio was counted using a hemocytometer (Beckman Coulter) under a phase-contrast microscope (Nikon, Tokyo, Japan). According to the result, the cell suspension was centrifuged (230 × g for 8 min) and resuspended in an appropriate volume of DMEM supplemented with 10% FBS (without extract D) to yield a final cell concentration of 106/ml. The suspension was then transferred into six-well plates (1 ml per well; Nunc) and incubated in a CO2 incubator at 37°C. After 24 h, 1 ml of prepared DMEM containing 0.125, 0.25, 0.5, 1, and 2 mg/ml of extract D was added to five of the wells to final concentrations of 0.0625, 0.125, 0.25, 0.5, and 1 mg/ml, respectively, and the final well served as a control, to which 1 ml medium without extract D was added. The plate was maintained in the incubator for 5 days. Each day, 50 μl of supernatant was aspirated from each well, labeled, and preserved at 4°C for further spectrometric test.
Cell Count
At the end of the fifth day, after the last 50 μm of supernatant was drawn from each well, all the residual media were removed, and the wells were rinsed with PBS twice. One milliliter of 0.25% trypsin was added into each well. After 5 min of digestion, another 1 ml DMEM containing 10% FBS was added into each well, and the cells were mixed thoroughly. Cell suspension (50 μl) was aspirated from each well for cell count analysis using the Beckman Z2 Cell Counter (Beckman Coulter). Each specimen was tested three times, and the mean value was recorded. Inhibition rate was obtained using the following formula: inhibition rate (%) = [(number of cell count of control group– number of cell count of experimental group)/number of cell count of control group] × 100%.
Collagen Content Determination
All the supernatant collected for collagen content determination during the previous 5 days were treated at the same time. Altogether, 30 pieces of specimens were obtained from one skin sample. A spectrometer (Ultrospec 3300pro; Amersham Pharmacia, Little Chalfont, Buckinghamshire, UK) was used to detect the concentration of collagen according to the method described by Huszar et al. (7). Measurements were repeated three times for each specimen. Mean values of the data from eight skin samples obtained at the same time point under the same extract D concentration were calculated. Collagen concentrations in different groups were analyzed by repeated-measures analysis of variance followed by post hoc LSD test for comparison of the effect of extract D on collagen secretion using SPSS 13.0. Differences were considered statistically significant when p < 0.05.
In Vivo Growth Investigation of Cultured Human Fibroblasts
The fibroblast cell line used for this part of study was established from a skin sample obtained from the upper eyelid of a 28-year-old female. Subcultured cells at passage 5 were harvested and divided into three equal parts. One part was resuspended with PBS at a concentration rate of 2 × 107 ml and then transferred into 1-ml syringes (B. Braun Medical, Shanghai, China). The other two parts were frozen for subsequent injections. The injections were performed subcutaneously with 1 ml cell suspension each time on the left side of the abdomen from cephalad to caudal (the medial part of the forelimb, the central part of the abdomen, and the medial part of the hindlimb) at 1-month intervals. For all the mice, the corresponding sites on the right abdomen were used as blank controls, where 1 ml PBS was injected at the same time the fibroblasts were injected into the corresponding left site. Each injection site was marked by a tattoo spot. At the end of the third month, all the injection sites were biopsied together so as to obtain the tissue specimens 1, 2, and 3 months after injection simultaneously. All the tissue specimens were fixed, dehydrated, embedded in paraffin (Yili Chemicals), and made into 4-μm sections for further hematoxylin and eosin (H&E; Yili Chemicals) and immunohistochemical staining.
H&E Stain
H&E stain was performed according to the common protocols.
Immunohistochemical Stain
After deparaffinization, the sections were put into 3% H2O2 (Yili Chemicals) for 8 min to deactivate the peroxisome. After rinsing with H2O2, they were incubated in primary antibodies [rabbit monoclonal antibodies against human type I collagen (Boster, Nanjing, China) and rabbit monoclonal antibodies against human type III collagen (Zhongshan Goldenbridge, Beijing, China)] for 1 h at 37°C. Secondary antibody (biotin-conjugated goat anti-rabbit monoclonal antibodies, Boster) was then added, and the sections were incubated at 37°C. Twenty minutes later, horseradish peroxidase-labeled streptavidin (SA/HRP; Zymed Laboratories, S. San Francisco, CA, USA) was added, and the sections were incubated for another 20 min at 37°C. The sections were washed three times in PBS and then incubated in diaminobenzidine (DAB; Zymed Laboratories). After color developed, the sections were examined under UV illumination and photographed. PBS was used in replacement of primary antibody as negative control.
Results
Genomic Stability of In Vitro Cultured Human Fibroblasts
Flow Cytometry
Table 1 and Figure 2 show the results of flow cytometry. Compared with passages 5 and 10 using Wilcoxon matched-pairs signed-ranks test, p values of G1, G2, S, and G2/G1 were 0.327, 0.889, 0.208, and 0.483, respectively, suggesting that there were no significant differences in cell cycles between the two passages.

Cell cycle analysis of sample 3. Propidium iodide staining was performed, and the proportion of cell cycle phase (measured as DNA content) was analyzed by flow cytometry. (A) Passage 5. (B) Passage 10.
Flow Cytometry Data at Different Passages and Phases of the Cell Cycle

Karyotype Analysis
All the cells chosen from the eight patients were normal 46, XX or 46, XY at both passages 5 and 10. No obvious mutations were found (Fig. 3).

Karyotype analysis of sample 3. (A) Passage 5. (B) Passage 10. No obvious mutations were found.
Growth Regulation Test at the Presence of Skin Extracts
Collagen Concentration
Table 2 and Figure 4 show the effect of different concentrations of extract D on collagen secretion of the cultures. Although collagen concentration gradually increased in all of the six groups, in general, a decreased collagen content was observed associated with an increased concentration of extract D at the same time point. Repeated measures analysis of variance showed an overall significant effect of the extract D concentration on collagen secretion over 5 days (F = 3.206, p = 0.015). Post hoc test showed that the inhibitory effect of extract D was significant at or above 0.125 mg/ml (p < 0.05). However, no significant differences were found between the four groups in which the extract D concentration was at or higher than 0.125 mg/ml.

Effect of different concentrations of extract D on collagen secretion.
Collagen Concentrations Under Different Concentrations of Extract D at Different Time Points (Mean ± SD; μg/ml)
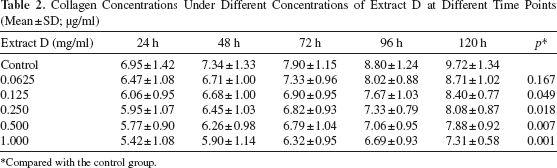
Compared with the control group.
Inhibition Rate
Table 3 and Figure 5 show the different cell counts and inhibition rates under the different concentrations of extract D at the end of day 5. The cell quantity was inversely proportional to the concentration of extract D, and the inhibition rate was directly proportional.

Cell counts and the inhibition rates under different concentrations of extract D at the end of day 5.
Cell Counts and Inhibition Rates Under Different Concentrations of Extract D at the End of Day 5

In Vivo Growth Investigation of Cultured Human Fibroblasts
Gross Morphology
The general condition of every nude mouse in the study was good without accident, death, or wound infections. The injection sites appeared normal; no obvious erythema, swelling, or ulcer was discovered. The skin was soft and normal, harmonious with the surrounding skin; no prominence or nodule was found during palpation in both experimental and control sites.
H&E Stains (Figs. 6 and 7)
On the control sites, the density of fibroblasts was remarkably lower than that on the experimental sites. Collagen fibers arranged regularly and parallel to the skin surface. Fibroblasts showed typically spindle and elongated shape. However, on the experimental sites, the number of the cells increased gradually after injection. These fibroblasts were first concentrated along the superficial dermis where they were injected and then proliferated and distributed throughout the dermis. Regularly arranged collagen bundles were interrupted by the injected cells. The nuclei of the cells were big, spherical, and deep stained at first and then became round and darker. Mitotic cells with the typical double-nucleolus presentation were easily observed at the end of the first month but were rare at the end of the second and third months. Most of the fibroblasts were confined in the dermis. There were no obvious macrophages or fibrous capsule around. No dysplastic cells were found.
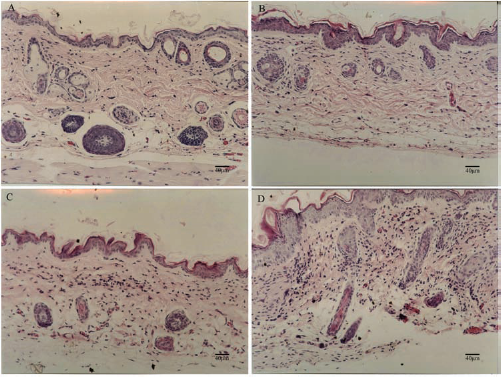
Biopsy specimens of the control site 3 months after injection (A) and the experimental sites 1 month (B), 2 months (C), and 3 months (D) after injection (H&E, 10 × 5). The density of fibroblasts was remarkably lower on the control side than that on the experimental side. On the experimental side, the number of the cells increased gradually after injection. Scale bars: 40 μm.
Biopsy specimens of the control site 3 months after injection (A) and the experimental sites 1 month (B) and 3 months (C) after injection (H&E, 100 × 5). Fibroblasts showed typically spindle and elongated shape on the control site. However, on the experimental sites, fibroblasts with big spherical and deep-stained nucleus were discovered. The nucleus became round and darker as the time went by. Mitotic cells with the typical “double nucleolus” were easily observed at the end of the first month, but rare at the end of the second and third months. Scale bars: 5 μm.
Immunohistochemical Stain (Figs. 8 and 9)
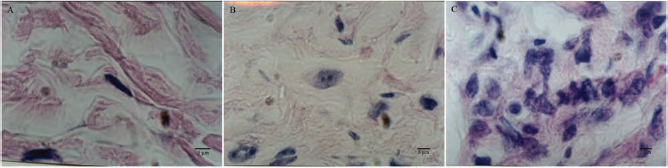
At the end of the first month, no obvious type I collagen stains were found around the cells; however, type III collagen-positive cells were clearly observed. At the end of the second month, both collagens were discovered inside as well as outside the cells. More positive cells were observed, and the stains became darker at the end of the third month. Intracellularly, under high-magnification microscope, type I collagen mostly located peripherally near the membrane, whereas type III collagen was observed filling the cell, almost covering the nucleus. Extracellularly, an increased density and thickness of both collagens were seen as the time went by. Absence of staining was shown in both blank and negative controls, confirming the specificity of the antibodies.

Immunohistochemical stain of type I and type III collagens. At the end of the first month, no obvious type I collagen stains were found around the cells (A, 20 × 5). However, type III collagen-positive cells were clearly observed (E, 20 × 5). At the end of the second month, both type I (B, 20 × 5) and type III (F, 20 × 5) were discovered inside as well as outside the cells. At the end of 3 months, more positive cells of type I (C, 20 × 5) and type III (G, 20 × 5) collagens were observed, and the stains became darker. Intracellularly, type I collagen was mostly located peripherally near the membrane (D, 60 × 5), whereas type III collagen was observed throughout the cell, almost covering the nucleus (H, 60 × 5). Scale bars: 20 μm (A, B, C, E, F, G) and 10 μm (D, H).

Negative controls of type I (A) and type III (B) collagens and blank controls of type I (C) and type III (D) collagens 3 months after injection (20 × 5). Scale bars: 20 μm.
Discussion
An ideal filler substance to correct wrinkles and scars should attain the following requirements: good biocom-patibility, permanence, minimal side effects, and easy to harvest and use for the physicians. Over the past several decades, numerous attempts have been made to develop such a product. Autologous materials such as autologous fat may obviate the risk of hypersensitivity responses caused by allogeneic, xenogeneic, or artificial materials. Unfortunately, it also has limitations, such as overcorrection, easy absorption, and inability to treat the dermis.
The use of autologous fibroblasts for dermal defect augmentation represents a potentially exciting natural alternative to the use of other filler materials, which meets all the above-mentioned requirements. Although phase III clinical trials and histological analysis have demonstrated sustained clinical improvement by increased collagen formation and dermal thickness (2,3,8,17,18), proper preclinical investigations are necessary to identify the growth character and to ensure biological safety and functional activity of fibroblasts both before and after transplantation.
Clinically, cells at passages 3–4 are most suitable for injection in terms of the cell quantity and their proliferative and secretory activity. We chose the cells at passage 5 for genomic detection to ensure the stability of the cells at previous passages. Besides, Hayflick and Moorhead (5) have shown that human fibroblasts could maintain their genomic stability after 40 generations, although cells at the 10th passage (20th to 30th generations) were too senescent for injection. Thus, for safety concerns, cells at passage 10 were also chosen to further confirm the genetic stability. Flow cytometric cell cycle analysis and karyotype analysis were employed to prove genomic stability from both global and individual perspectives. Flow cytometric cell cycle analysis at 95% confluence showed no difference in the fraction of cells in the G1/G0, S, and G2/M phases between passages 5 and 10. No hypodiploid peak or hyperdiploid peak was observed, and there was no significant change in the distribution of G1/G0, S, and G2/M fractions, which indicated that generally the proliferative behavior of the cultured cell population remained stable from passages 5 to 10 without overactive division or apoptosis. Classic karyotype analysis was performed to investigate the genomic stability of individual cell. In all the samples, both cells at passages 5 and 10 maintained their basic diploid karyotype consistent with normal somatic cells. Mutations or other translocations were not discovered at either passage 5 or 10. Both flow cytometric and karyotype analyses showed that, in general, no chromosomal abnormalities were found in in vitro expanded human fibroblasts. However, because of the low-resolution capacity of our approaches, we could not completely exclude small segment mutations or other subtle molecular events. Telomerase activity and carcinogenicity should be measured in a further study to eliminate tendency of malignant transformation.
Scar formation and contraction are two things that should be avoided when using fibroblast treatment. In case of correction dermal and subcutaneous depression, where the skin remains intact, the possibility of wound contraction is very low. That leaves fibrosis and scar formation as our primary concern, which may be caused by excessive growth or secretion of cultured fibroblasts after transplantation. Previous studies have shown that the extracts of the dermis (extract D) could inhibit the proliferation of fibroblasts (4,11). With the intention to confirm that there did exist something in normal skin that regulated the growth of fibroblasts, we repeated the in vitro experiments with some modifications. The results showed that the decreased collagen concentrations and cell counts were correlated with an increased concentration of extract D. The extract D showed a significant inhibitory effect at or above 0.125 mg/ml. However, the relationship with extract D and the secretion of the fibroblasts required further investigation. The main component of extract D was discovered by Stewart (15) as dermatan sulfate proteoglycan II (decorin), which has recently been confirmed to have a downregulatory effect on cell proliferation (14,19). Although its in vivo concentration in normal skin and the exact mechanism are still not clear, we are now planning to study the expression of decorin in the injected area to see if it really helps in regulating the injected fibroblasts.
In our previous study, cultured autologous skin fibroblasts had been proven to survive for at least 5 months after injection (20). In this study, cultured human skin fibroblasts were injected into nude mice to further demonstrate their proliferation and secretion function. The injected human fibroblasts could be easily distinguished from the original mouse fibroblasts because of their oval shape and big spherical nucleus. They appeared to be incorporated into the dermal architecture and began to secrete collagen. Good vitality of the injected cells could be inferred from both the increase in cell number and the existence of mitotic cells. On the other hand, the proliferation was active at the first month and returned to normal later, indicating that the proliferation of the injected cells was under certain regulations, which prevented the cells from hyperplasia and ensured the safety of the transplantation. The fact that no macrophages were found suggested that there were no abnormal apoptosis or necrosis of the injected cells, which also confirmed not only the vitality of these cells but also the safety of this procedure. Moreover, normal cell morphology without dysplasia was observed from histological sections, and no tumors were detected from gross inspection together, suggesting that no oncogenic transformation or fibrosis formation had occurred at least 3 months after transplantation.
As for their secretory function, fibroblasts secrete different kinds of extracellular matrix proteins, of which collagen is the one we expect to be most likely involved in correcting dermal and subcutaneous defects. Type I and type III collagens are the most abundant types of collagens in the skin, which are responsible for the skin's mechanical properties. In adults, type I collagen constitutes approximately 80% of dermal collagen, whereas type III collagen is abundant in fast growing tissue, particularly at the early stages of wound repair, and then gradually replaced by the stronger and tougher type I collagen as the wound repair process slows down (10). In our study, both types of collagens were observed. Using immunohistochemical staining, these collagens were demonstrated to be secreted by the transplanted human fibroblasts. The fact that the reaction of type III collagen appeared earlier and stronger than that of type I collagen after injection was consistent with their different physiological functions. Both types of collagens accumulated gradually during the 3 months. At the end of the third month, round- or oval-shaped fibroblasts could still be observed with positive reactions for either type of collagen deposited inside, suggesting an active and long-lasting secretory function of the cells, which would provide a continuous corrective effect.
Despite the above findings, the in vivo section of the study still has limitations. First, since the injected cells were not labeled, it is hard to know their behavior after transplant. Second, the viability and stability of the injected cells were only demonstrated by microscope observation, which is not sufficient and so needs to be further clarified. Third, 3 months might not be long enough to prove a long-lasting and continuous result. In our future studies, long-term observation is going to be performed at both macro- and microlevels. In vivo cell tracking is planned to be used to understand their overall in vivo behavior and assess their migration and metastatic potential. At the cellular level, viability and genomic stability tests of the injected cells are going to be performed to quantitatively elucidate the safety and effectiveness of the technique.
From our preclinical study, the use of cultured human skin-derived fibroblasts was proven to be safe, providing a basic support for clinical use of autologous fibroblasts transplantation.
Footnotes
Acknowledgments
We are grateful to Professor Fude Fang Institute of Basic Medical Sciences, Peking Union Medical College (PUMC), and Chinese Academy of Medical Sciences (CAMS) for their help and advice. The study was supported by the National Natural Science Foundation of China. The authors declare no conflict of interest.