
Abstract
In this study, we used human amniotic membrane (AM) to prepare a dermal scaffold with intact basement membrane (BM) and good biostability for quick expansion and transplantation of epidermal keratinocytes (EKs). Fresh AM was treated by repeated freeze–thaw cycles and DNase digestion. This new method was able to cleanse the cell components effectively and retain the BM structure with continuous distributions of laminin, collagen IV, VI, and VII. Subsequently, the acellular amniotic membrane (AAM) was cross-linked with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) for 5 min, 30 min, and 6 h. With the time of cross-linking prolonging, the mechanical strength and biostability of AAM increased gradually, while its cytotoxicity to EKs also increased. The 5-min cross-linked AAM (5min-AAM) had no significant cytotoxicity with good histocompatibility. The relative cell viability of EKs seeded on the 5min-AAM surface was 367 ± 33% and 631 ± 43% at 7 and 14 days of culture, respectively, both higher than 294 ± 30% and 503 ± 41% of the conventional cell culture dish (CCD) group, and the proportion of P63-positive cells was significantly higher than that of the CCD group on day 7 (54.32 ± 4.27% vs. 33.32 ± 3.18%, p < 0.05). When the 5min-AAM loaded with EKs (EK-AAM) was grafted onto full-thickness skin defects in nude mice, the cells survived well and formed an epidermis similar to normal skin. The new epidermis was thicker, and reconstruction of the dermal structure was good with an intact BM. Four weeks after transplantation, the wound contraction rate in the EK-AAM group was 43.09 ± 7.05%, significantly lower than that in the EK sheet group (57.49 ± 5.93%) and control group (69.94 ± 9.47%) (p < 0.05). In conclusion, repeated freeze–thaw treatment with appropriate EDC cross-linking offers AAM an intact BM structure with good operability and biostability. It may prove to be an ideal dermal scaffold to promote expansion of EKs in vitro and be transplanted for reconstruction of the dermal structure.
Keywords
Introduction
How to maintain the proliferating activity of human epidermal keratinocytes (EKs) on the surface of dermal scaffolds is the key for survival of tissue-engineered skin substitutes. EKs will differentiate continuously and lose their proliferative ability gradually during the process of proliferation on the surface of the dermal scaffold, and only a small portion of the basal cells can maintain their high proliferative activity. The main reason is that the dermal scaffold lacks basement membrane (BM) structure and a continuous loss of epidermal stem cells during the process of culture (2, 24, 26). Although some currently available dermal scaffolds such as collagen and hyaluronic acid hydrogel (11, 33, 42), biodegradable electrospun scaffolds (4), and acellular matrices (24, 46) can well mimic the dermal structure, they all lack effective BM structures. Addition of certain natural or synthetic BM components to the dermal scaffold surface has been shown to promote the proliferation of EKs (13, 32, 38). Allogeneic/heterogeneic acellular dermis contains part of natural BM components, which can benefit adhesion and proliferation of the keratinocytes (24, 30). However, strong decellularization detergents and long time of processing are needed to remove the epidermis layer and vascular endothelial cells in the dermal matrix, and these treatments cause varying degrees of damage to the BM structure (5, 34). Therefore, more and more studies have focused on constructing dermal scaffolds with BM structures.
Amniotic membrane (AM) comprises a single layer of epithelial cells, a dense BM, and an avascular stromal matrix rich in collagen. The amniotic matrix mainly comprises fibroblasts without containing capillaries or vascular endothelial cells (28, 36). The BM layer is composed of laminin and collagen IV, VII, and XVII. It is the thickest BM in human body and is morphologically similar to the dermal and corneal BM (6, 28). Acellular amniotic membrane (AAM) matrix with BM structure has been proved to be a favorable substrate for expansion of human limbal stem cells, mesenchymal stem cells, and epidermal stem cells, preventing cell differentiation and maintaining the properties of stem cells (12, 20, 39); thus, it could be a good carrier candidate for quick proliferation and grafting of EKs. However, the currently available decellularization methods always cause varying degrees of damage to the BM structure (10). In addition, it is difficult to handle AM because it is very soft with low mechanical strength and biostability, and grafted AM is likely to degrade quickly (22, 43), which limit its practical use.
In the present study, we used repeated freeze–thaw cycles and DNase digestion to prepare an AAM matrix with intact BM structure and cross-linked it with soluble 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) to improve its mechanical strength and biostability. The cross-linked AAM thus obtained was able to promote the adhesion and proliferation of EKs markedly, and promote epidermis formation and dermal reconstruction when grafted to full-thickness skin defects.
Materials and Methods
Preparation of Acellular Amniotic Membrane
The study protocol was approved by the Ethics Committee of the hospital. AM was obtained from placentas of parturients who underwent cesarean section after obtainment of informed consent and exclusion of HIV, syphilis, HBV, and HCV infection. AM was rinsed and sterilized as previously reported (23), sheared into 4 × 4-cm chips, and randomly divided into two groups: (1) Freeze– thaw + DNase AM group, where AM was placed in 1.5-ml centrifuge tubes (Sigma-Aldrich, St. Louis, MO, USA), tranquilized in liquid nitrogen for 30 min, and placed in water bath at 25°C for 10 min. This procedure was repeated for two to five times to determine the optimal number of freeze–thaw cycles. Then, they were digested by DNase (1 mg/ml, Gibco, Gaithersburg, MD, USA) under gentle oscillation at 37°C for 3 h, and rinsed with phosphate-buffered saline (PBS; Sigma-Aldrich) three times, 3 min for each time. (2) Dispase II + scraping AM group, where AM was digested by dispase II (1.2 U/ml, Gibco) in Ca2+- and Mg2+-free Hanks' balanced salt solution (Beyotime, Jiangsu, China) at 37°C for 30 min, and then residual epithelial cells were scraped off gently with a cell brush (Corning cell scraper; Sigma-Aldrich) and rinsed thoroughly with PBS. Untreated intact AM was used as control.
Analysis of the Decellularization Effect
Histological Observation
AM specimens were routinely fixed with 4% paraformaldehyde (Pharmaceutical & Chemical Reagent Company, Shanghai, China), dehydrated, paraffin embedded, sliced into 5-μm sections, and stained with hematoxylin and eosin (H&E; Beyotime) for observation. Parts of the specimens were surface stained by H&E without slicing (45). The remaining specimens were incubated with Hoechst 33342 (DNA staining, 10 μg/ml, Beyotime) at 37°C for 15 min, washed twice with PBS, and observed for DNA residues under a fluorescence microscope (excitation: 350 nm, emission: 461 nm; Leica 3000, Tokyo, Japan).
Immunohistochemical Staining
BM-related components (laminin, collagen IV, VI and VII), AM tissue-related antigens [major histocompatibility complex (MHC)-I and MHC-II], and vimentin expression were examined as previously described (10). Briefly, the paraffin sections were routinely dewaxed, rinsed, incubated with 4% fetal bovine serum (Gibco, Gaithersburg, MD, USA) for 1 h, and then incubated with corresponding monoclonal antibodies against MHC-I, MHC-II, laminin, collagen VI (1:200, AbCam, Cambridge, UK), collagen IV, vimentin (1:100, Bioworld Consulting Laboratories, Mt. Airy, MD, USA), and collagen VII (1:100, Santa Cruz Biotechnologies, Santa Cruz, CA, USA) at 4°C overnight. The primary antibodies were detected with IgG-fluorescein isothiocyanate (FITC) fluorescence (for BM-related components, 1:300, Santa Cruz) or horseradish peroxidase (HRP)-labeled antibodies (for MHC-I/II and vimentin, 1:300, Santa Cruz) and visualized with diaminobenzidine (Boster, Wuhan, China). Hoechst was used for counterstaining the nuclei. The slides were examined and photographed under a fluorescence microscope.
Preparation of Cross-Linked AAM
The AAM was placed in 30 ml of PBS with addition of EDC (0.05 mmol/mg AAM, Sigma-Aldrich, St. Louis, MO, USA) and N-hydroxysulfosuccinimide (NHS, 0.2 mol/ mol EDC, Sigma-Aldrich), and cross-linked on a thermostatic shaker (30 rpm; THZ-300; Heng Technologies Co. Ltd., Shanghai, China) at 37°C for 5 min, 30 min, and 6 h. After cross-linking, the specimens were rinsed thoroughly with 95% ethanol, ddH2O, and PBS. The extent of EDC cross-linking was determined by the ninhydrin method (22) and expressed as the cross-linking index, the percentage of free amino groups reacting with EDC in AAM. Non-cross-linked AAM and 5-min, 30-min, and 6-h cross-linked AAM were termed as 0min-AAM, 5min-AAM, 30min-AAM, and 6h-AAM, respectively. The experiment was repeated five times.
Analysis of the Characteristics of Cross-Linked AAM
Morphology, Mechanical Strength, and In Vitro Degradation Rate
The gross appearance of AAM before and after cross-linking was observed. The ultrastructure was examined by field emission scanning electron microscopy (SEM; QUANTA FEG 450; FEI, Hillsboro, OR, USA). The mechanical strength was measured with a uniaxial tension meter. Briefly, AAM specimens were hydrated in PBS for 24 h and sheared into dog bone-shaped pieces (gauge length, 12 mm; width, 6 mm). The maximum tension and the maximum stretch length were measured with an Instron tensiometer (Instron, Norwood, MA, USA) at the strain rate of 5 mm/min. The in vitro degradation rate of AAM was measured by type 1 collagenase (Clostridium histolyticum, Sigma-Aldrich) digestion method, that is, weight change was measured after 28-day digestion of the tested specimen in 24 μg/ml type 1 collagenase at 37°C to calculate the degradation rate = [(Wbefore – Wafter) / Wbefore] × 100%. The result was averaged from five independent runs.
Analysis of Cytotoxicity
AAM (1.5 × 1.5 cm) was placed at the bottom of a six-well plate (Corning, Corning, NY, USA) with the epithelial side upward and immobilized with stainless steel rings (inner diameter, 20 mm; in house). Human EKs were isolated from the foreskin of five healthy teenagers (aged 13–17) (35). Passage 3 EKs were seeded at 2 × 104 cells/cm2 and cultured in serumfree culture medium (Keratinocyte growth medium 2; PromoCell Company, Heidelberg, Germany) for 7 days, with the medium changed on alternative days. After 1, 3, 5, and 7 days of culture, 5-mm punch biopsies were collected and placed into separate wells in a 96-well plate (Corning). Cell viability was measured by standard Cell Counting Kit-8 (CCK-8, Beyotime) method. Briefly, each well was added with 100 μl of CCK-8 solution and cultured for 4 h. The solution was moved to a new 96-well plate to measure absorbance with a multiplate reader (Biotek, Winooski, VT, USA) at 450 nm. Cells were also stained with Hoechst 33342 and propidium iodide (PI) (Beyotime) to observe cell viability (excitation light: 355–488 nm, emission light: 460–615 nm). Using ImagePro Plus Software (Media Cybernetics, Rockville, MD, USA), the percentage of cells with different labels in nine random fields was measured in the 5min-AAM, 30min-AAM, and 6h-AAM groups, and the 0min-AAM group was used as control. Each group had five specimens.
Observation of Histocompatibility
All animal experiments were performed according to the NIH guidelines for animal care and use. Fifteen immune competent SD male rats weighing 180–200 g (Experimental Animal Center of the Second Military Medical University, Shanghai, China) were anesthetized with intraperitoneal (IP) injection of 1% sodium phenobarbital (Sigma-Aldrich). Cross-linked AAM (2×2 cm) was aseptically embedded subcutaneously on both sides of the dorsal midline (19). The materials were obtained 1, 2, 4, and 8 weeks and 3 and 4 months after surgery, with three specimens for each group at each time point. The tissues were processed for H&E and Masson's trichrome staining (Yuanye Biotechnology, Shanghai, China) to observe the in vivo degradation process of the cross-linked AAM. Inflammatory response and vascularization were observed by immunohistochemical staining of cluster of differentiation 11b (CD11b; labeling monocytes and granulocytes), CD68 (labeling monocytes and macrophages), CD4 (labeling helper T lymphocytes), CD8 (labeling cytotoxic T lymphocytes), CD31 (labeling vascular endothelial cells), and vimentin (labeling fibroblasts, endothelial cells and other mesenchymal cells). The staining procedures were the same as described above, using primary antibodies against CD11b, CD68, CD4 and CD8 (1:200, AbCam), CD31 (1:200, Santa Cruz), and vimentin (1:100, Bioworld), and diaminobenzidine was used for color development.
Expansion of Human EKs on Cross-Linked AAM
Passage 3 human EKs were seeded onto the cross-linked AAM at 5 × 104 cells/cm2 and cultured for 2 weeks to form EK-AAM. Cell proliferation viability was detected at days 1, 3, 5, 7, 9, 11, and 14 by standard CCK-8 method, and cell density was also observed by Hoechst staining. On day 14, the specimens were collected and processed for H&E staining. P63 (a nuclear transcription factor indicating the presence of undifferentiated proliferating cells) expression of the EKs was observed by immunohistochemical staining using primary antibody against P63 (1:150, Santa, USA) and IgG-FITC secondary antibody (Santa Cruz), and the percentage of P63-positive cells was calculated in nine randomly selected fields using Image-Pro Plus Software. EK sheet cultivated on a cell culture dish (CCD) (Corning, Corning, NY, USA) by the same procedures was used as the control. Five specimens were selected for each group.
Transplantation of EK-AAM to Full-Thickness Skin Defects
Thirty-six immunodeficient nude mice aged 6 weeks (male and female from B&K Universal Group Ltd.; Hull, UK) were anesthetized by IP injection of 1% sodium pentobarbital, and a 1.5-cm full-thickness skin defect was established on both sides of the dorsal midline. The animals were randomized to EK-AAM, EK sheet, and control groups (12 mice for each group). EK-AAM and EK sheet were spread flat over the wound surface, fixed on the skin around the wound by intermittent suturing with 4–0 silk thread (Ethicon, Johnson & Johnson, Shanghai, China), and covered with vaseline gauze (Medical Instrument Company, Shanghai, China). The control group was covered with vaseline gauze only. At 1, 2, 3, and 4 weeks after transplantation, photographs of the wounds were taken from a certain distance. The wound healing rate and wound contraction rate were calculated using Image-Pro Plus Software. Specimens were taken periodically and stained with H&E. The distribution of BM-related component laminin (1:200, AbCam, UK) was observed by immunohistochemical staining. The primary antibody was detected with IgG-FITC fluorescence. Hoechst was used for counterstaining the nuclei.
Statistical Analysis
All data were tested for normal distribution and homogeneity of variances by the Shapiro–Wilk and Levene methods. Measurement data of normal distribution and homogeneity of variance were expressed as mean ± standard deviation. Intergroup comparisons were performed by Student's t test (between two groups) or analysis of variance (among more than two groups, post hoc multiple comparisons were tested using SNK method). Analysis was performed using SPSS16.0 software (IBM, Armonk, NY, USA). A value of p < 0.05 was considered statistically significant.
Results
Preparation of AAM with Intact BM
A satisfactory decellularization effect could be achieved by three to five freeze–thaw cycles. To minimize damage to the BM structure, we finally chose the three freeze–thaw cycle protocol. Figure 1 shows the decellularization effect of different treatments. The cell membrane was broken after repeated freeze–thaw cycles, including AM epithelial cells and fibroblasts in the matrix, but the nuclei still existed (Fig. 1B, G), and after DNase digestion, the nuclei were also effectively removed (Fig. 1C, H). Hoechst superficial staining showed that only a small amount of nucleic acid remained (Fig. 1M). The epithelial cells and fibroblasts could not be completely removed by dispase II digestion alone, as residues of scattered epithelial cells and fibroblasts, or even masses of epithelial cells, were detected (Fig. 1D, I). The epithelial cells could be removed almost completely after additional scraping treatment; however, residues of fibroblasts were still visible in the matrix (Fig. 1E, J). Hoechst superficial staining showed that there were still moderate amounts of nucleic acid residues (Fig. 1O).
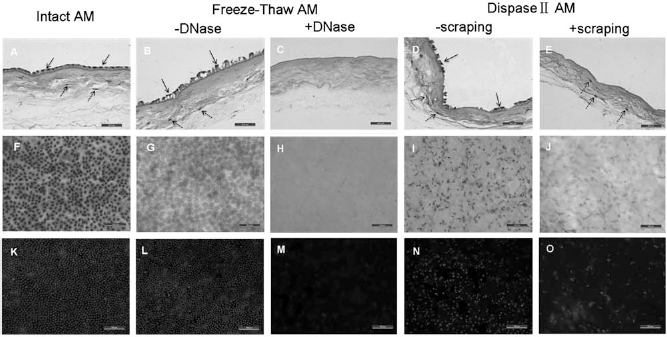
Decellularization of AM. (A–E) H&E staining of cross sections shows removal of amniotic membrane (AM) epithelial cells (solid arrows) and fibroblasts in the matrix (dotted arrows). (F–J) Superficial H&E staining shows the AM superficial structure and removal of epithelial cells. (K–O) Hoechst superficial staining shows removal of DNA in AM. The epithelial cells and fibroblasts are effectively removed from AM by repeated freeze–thaw and DNase treatment (C), while dispase II digestion and scraping treatment cannot remove the cells completely (dotted arrows in E). Scale bars: 50 μm (A–J); 100 μm (K–O).
Immunohistochemical staining showed that laminin, collagen IV, VI, and VII continuously distributed at the BM area of AM treated by repeated freeze–thaw cycles and DNase digestion, and collagen IV and VI were also seen in the stroma as observed in the intact AM. These BM-related proteins almost disappeared after dispase II digestion and scraping treatments, and the density of collageIV and VI in the stroma also decreased markedly (Fig. 2).
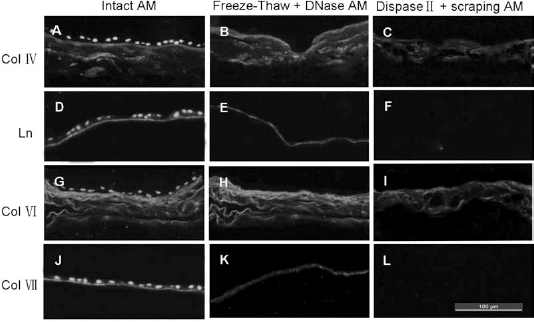
Immunohistochemical observation of the distribution of BM-related proteins in treated AM. Immunohistochemical observation of the distribution of the basement membrane (BM)-related proteins (laminin, collagen IV, VI, and VII) in the treated AM groups; intact AM (left panels), freeze–thaw + DNase AM (middle panels), and dispase II + scraping AM (right panels). The nucleus is stained by Hoechst. Collagen IV (A–C), laminin (D–F), collagen VI (G–I), and collagen VII (J–L) are all seen uniformly distributed over the intact and freeze–thaw + DNase AM; however, they are mostly missing in the dispase II + scraping AM.
Immunohistochemical staining of MHC-I, MHC-II, and vimentin showed that both the AM epithelial cells and fibroblasts expressed MHC-I antigen (Fig. 3A) and did not express MHC-II antigen (Fig. 3B), and only the fibroblasts expressed vimentin (Fig. 3C). The stainings of MHC-I (Fig. 3D), MHC-II (Fig. 3E), and vimentin (Fig. 3F) in the AAM treated by repeated freeze–thaw and DNase digestion were all negative, indicating that both the AM epithelial cells and fibroblasts in the matrix were removed completely, as well as their related antigens.

Immunogenicity of acellular amniotic membrane (AAM). (A–C) Immunohistochemical staining of major histocompatibility complex (MHC)-I, MHC-II, and vimentin in intact AM, respectively. Both AM epithelial cells (solid arrows) and fibroblasts (dotted arrows) express MHC-I antigen (A) and do not express MHC-II antigen (B), and only the fibroblasts express vimentin (dotted arrows in C). (D–F) Immunohistochemical staining of MHC-I, MHC-II, and vimentin in AAM after repeated freeze–thaw and DNase treatment, respectively. All the stainings are negative, indicating that both AM epithelial cells and fibroblasts in the matrix have been removed completely, as well as the related antigens. Scale bars: 50 μm.
Preparation of Cross-Linked AAM
After EDC cross-linking for 5 min, 30 min, and 6 h, the cross-linking index of AAM was 55.56 ± 2.84%, 82.28 ± 1.44%, and 93.62 ± 1.09%, respectively, and the difference was significant between the groups (p < 0.01). The 0min-AAM was soft, smooth, and difficult to handle as it was easy to coil up (Fig. 4A). The 5min-AAM was a bit harder with a flat and smooth surface and was easy for handling (Fig. 4B). However, as the cross-linking time further prolonged, the AAM became rough, hard, and stiff. The 6h-AAM coiled badly with poor plasticity, and it was difficult to spread it over the dish (Fig. 4C). SEM showed that EDC cross-linking made the original uniform and dense mesh collagen fibers of the 0min-AAM associate with each other into thicker fibrous bundles or even cords (Fig. 4D–F).

Changes over time in the gross appearance and SEM images of cross-linking AAM. Gross appearance and scanning electron microscope (SEM) images of 0min-AAM (left panels), 5min-AAM (middle panels), and 6h-AAM (right panels). As cross-linking time prolongs, the soft and smooth AAM gradually turns into a coiled and stiff one (A–C), and the uniformly reticular structure of the collagen fibers is replaced by fibrous cords formed by cross-linking (D–F).
The mechanical strength of AAM is represented as the maximum tension and the maximum stretch length. The maximum tension of the 5min-AAM, 30min-AAM, and 6h-AAM was 3.07 ± 0.25 N, 3.77 ± 0.21 N, and 4.14 ± 0.31 N, respectively, all of which were higher than 1.42 ± 0.15 N of the 0min-AAM (p < 0.01). The maximum stretch length of the 5min-AAM, 30min-AAM, and 6h-AAM was 5.82 ± 0.64 mm, 4.91 ± 0.30 mm, and 4.07 ± 0.40 mm, respectively, all of which were lower than 12.87 ± 0.96 mm of the 0min-AAM (p < 0.01). The mechanical strength of AAM increased and the stretch length decreased with the extent of cross-linking increase.
The 0min-AAM was degraded completely after 7 days of collagenase digestion, while the degradation rate of the 5min-AAM, 30min-AAM, and 6h-AAM was 45.83 ± 1.46%, 14.90 ± 1.45%, and 5.87 ± 0.97% at day 7, and 47.73 ± 0.85%, 16.80 ± 1.31%, and 7.43 ± 1.20% at day 28, respectively, all of which were significantly lower than that of the 0min-AAM at the same time point (p < 0.01). EDC cross-linking increased the biostability of AAM.
Cytotoxicity test showed that there was no significant difference in OD value between the 0min-AAM and 5min-AAM groups for 7 days (p > 0.05). The OD value of the 30min-AAM group was lower than that of the 0min-AAM and 5min-AAM groups at day 7 (1.70 ± 0.09 vs. 2.20 ± 0.07 and 2.18 ± 0.05, p < 0.01). EKs in the 6h-AAM group proliferated slowly, and from day 5 on, its cell viability was lower than that of the 0min-AAM group (1.15 ± 0.09 vs. 1.57 ± 0.06, p < 0.01), and at day 7, the OD value was significantly lower than that of the other three groups (1.30 ± 0.05 vs. 2.20 ± 0.07, 2.18 ± 0.05, and 1.70 ± 0.09, p < 0.01) (Fig. 5A). After live/dead staining, the nuclei of viable cells were light blue; apoptotic cells were bright blue with pyknotic nucleus; and dead cells were red (Fig. 5B–E). The percentage of apoptotic and dead cells in the 0min-AAM and 5min-AAM groups was 2.42 ± 0.34% and 2.44 ± 0.38%, respectively, without significant difference between them (p > 0.05). The percentage of apoptotic and dead cells in the 30min-AAM and 6h-AAM groups was 5.27 ± 0.50% and 10.02 ± 1.43%, respectively, both higher than that in the 0min-AAM and 5min-AAM groups (p < 0.05).

Cytotoxicity analysis of cross-linked AAM. (A) Cell viability of epidermal keratinocytes (EKs) cultured on different AAM for 7 days, detected by Cell Counting Kit-8 (CCK-8) assay. There is no significant difference in OD value between the 5min-AAM and 0min-AAM groups, while the OD value of the 30min-AAM and 6h-AAM groups begin to decrease at day 7 and 5, respectively, compared with the 0min-AAM group (*p < 0.01, n = 5). (B–E) Live/dead staining using Hoechst 33342 and propidium iodide (PI) shows that the nuclei of viable cells are light blue (ordinary arrows), apoptotic cells are condensed and bright blue (square-tail arrows), and dead cells are red (round-tail arrows). There are rarely apoptotic and dead cells in the 0min-AAM (B) and 5min-AAM groups (C), while the number of apoptotic and dead cells increased significantly in the 30min-AAM (D) and 6h-AAM groups (E). Scale bars: 200 μm.
In Vivo Histocompatibility of Cross-Linked AAM
Of all cross-linked AAM groups, only the 5min-AAM retained the flat and smooth morphology and the ability of supporting the growth of EKs as did 0min-AAM. Figure 6 shows the in vivo degradation process of 5min-AAM. The 5min-AAM's morphology remained almost unchanged 1 week after implantation, without evident acute inflammatory response or rejection reaction (Fig. 6A, D), and it degraded partially 4 weeks after implantation (Fig. 6B, E). The 5min-AAM almost degraded completely after 4 months, and a thick subcutaneous tissue formed, where new collagen was well arranged with abundant vessels, without evidence of space and cyst formation (Fig. 6C, F).
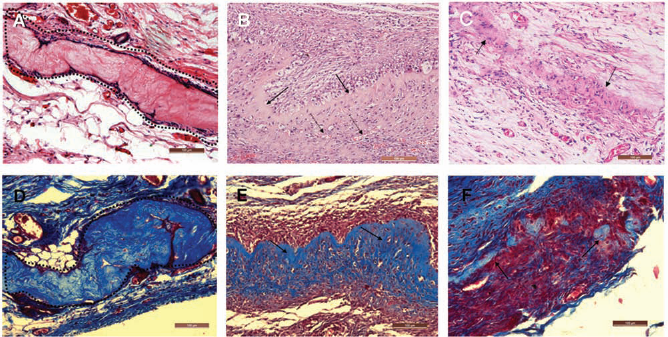
In vivo degradation of 5min-AAM. (A–F) H&E and Masson's trichrome staining results 1 week, 4 weeks, and 4 months after subcutaneous embedding of the 5min-AAM, respectively. (A, D) The 5min-AAM structure is almost unchanged after 1 week (dotted lines), without causing evident inflammatory response. (B, E) The 5min-AAM (solid arrows) degrades slowly, and invasion of new blood vessels (dotted arrows) can be seen. (C, F) The 5min-AAM is degraded almost completely after 4 months, forming a thick subcutaneous tissue. Scale bars: 100 μm.
The inflammatory response and vascularization of the 5min-AAM are shown in Figure 7. Immunohistochemical staining results showed that there was only a small number of cells positive for CD11b, CD68, CD4, and CD8 on week 4 after subcutaneous embedding, which means there was only a small amount of neutrophils, monocytes, macrophages (Fig. 7A, B), and T lymphocytes (Fig. 7C, D). Staining of CD31 and vimentin indicated the formation of many vessels and the invasion of large numbers of fibroblasts and other mesenchymal cells (Fig. 7E, F). These results show that there was less infiltration of inflammation-related cells after subcutaneous implantation of 5min-AAM, and most of the cells were fibroblasts, endothelial cells, and other mesenchymal cells.

Determination of in vivo histocompatibility. (A–D) Immunohistochemical staining of cluster of differentiation 11b (CD11b), CD68, CD4, and CD8 in the 5min-AAM after subcutaneous implantation for 4 weeks, respectively. There was only a small number of neutrophils, monocytes, macrophages (arrows in A, B), and T lymphocytes (arrows in C, D). (E, F) Immunohistochemical staining of CD31 and vimentin, respectively, indicating the formation of many blood vessels (arrows in E) and infiltration of large numbers of fibroblasts and other mesenchymal cells (arrows in F). Scale bars: 50 μm.
Quick Proliferation of Human EKs
The EKs adhered to the surface of 5min-AAM quickly and grew to confluence in 3 days. The cells were morphologically uniform and spindle-shaped. CCK-8 detection results showed that the relative cell viability of the 5min-AAM group at days 7 and 14 was 367 ± 33% and 631 ± 43%, respectively, versus 294 ± 30% and 503 ± 41% of the CCD group (p < 0.05) (Fig. 8A). The EKs on the 5min-AAM formed a two- to three-layer structure after being cultured for 14 days (Fig. 8B). On day 7, the percentage of P63-positive cells was 54.32 ± 4.27%, significantly higher than 33.32 ± 3.18% of the CCD group (p < 0.05) (Fig. 8C–E).

Comparison of EK proliferation between 5min-AAM and CCD groups. (A) The result of CCK-8 assay shows that the relative cell viability in the 5min-AAM group is significantly higher than that in the cell culture dish (CCD) group from days 7 to 14 (*p < 0.05, n = 5). (B) H&E staining of EK-AAM 14 days after culture. EKs form a two- to three-layer structure on the cross-linked AAM. Scale bar: 50 μm. (C) Hoechst fluorescence staining of EKs 7 days after culture in the 5min-AAM and CCD groups. Scale bar: 200 μm. (D, E) Immunohistochemical staining of P63 in EKs cultured on 5min-AAM and CCD, respectively. Scale bars: 100 μm.
Transplantation of EK-AAM to Full-Thickness Skin Defects
Both the EK-AAM and EK sheet survived well. One week after transplantation, the surviving cells in the EK-AAM and EK sheet expanded externally, and the wound surface was moist, while dry scabs formed in the control groups. The wound contraction rate in the EK-AAM, EK sheet, and control groups was 22.03 ± 5.48%, 21.87 ± 8.01%, and 24.84 ± 8.04%, respectively, without significant difference among them (p > 0.05). Two weeks after grafting, new epidermis in the EK-AAM and EK sheet groups became thicker gradually. The wound surface was dry, and only small residual wounds were seen in the margin. In the control group, the wound healed mainly by contraction with a residual wound in the center. The healing rate of the EK-AAM, EK sheet, and control groups was 92.0 ± 2.8%, 93.3 ± 2.8%, and 93.1 ± 5.2%, respectively, without significant difference among them (p > 0.05), but the wound contraction rate was 34.79 ± 7.29%, 41.19 ± 8.72%, and 59.08 ± 10.79%, respectively, indicating that wound contraction in the EK-AAM and EK sheet groups was less severe than that in the control group (p < 0.05). After 4 weeks, all the wounds healed completely. The epidermis in the EK-AAM group was soft and smooth, similar to normal skin, and the wound contraction rate was significantly lower than that in the EK sheet and control groups (43.09 ± 7.05% vs. 57.49 ± 5.93% and 69.94 ± 9.47%, p < 0.05) (Fig. 9).

Healing process of full-thickness skin defects. The wounds in the EK-AAM and EK sheet groups are covered with thin layers of epidermis 1 week after grafting, and the epidermis gradually becomes thicker after 2 weeks, with small wounds in the margin. The wounds in the control group heal mainly by contraction, and a small residual wound is seen in the center on week 2. The wounds heal completely in all three groups after 4 weeks. The epidermis in the EK-AAM group is flat and soft, similar to normal skin. The wound contraction is milder than that in the EK sheet and control groups.
H&E staining showed that 4 weeks after grafting the new epidermis in the EK-AAM group was thicker than that in the EK sheet and control groups. Increased amounts of cells were seen in the basal layer and were in orderly polarization arrangement in the conjunction between the epidermis and dermis. The 5min-AAM closely attached to the wound, with new capillaries growing into it, and the new collagen was well-arranged (Fig. 10A–C). Immunohistochemical staining of laminin showed that the BM formed in the EK-AAM group was thick and continuously distributed in the conjunction between the epidermis and dermis. On the contrary, the distribution of BM in the EK sheet and control groups was thin and intermittent (Fig. 10D–F).

Histological analysis of wounds 4 weeks after transplantation. (A–C) H&E staining shows that the newly formed epidermis in the EK-AAM group is thicker than that in the EK sheet and control groups. (D–F) Immunohistochemical staining shows that the distribution of laminin is continuous in the EK-AAM group, while it is intermittent in the EK sheet and control groups. Scale bars: 100 μm.
Discussion
Preparation of various kinds of dermal scaffolds is of significant importance in skin tissue engineering. Dermal scaffolds such as collagen hydrogel, biodegradable electrospun scaffolds, and acellular matrices can well mimic the dermal structure. However, since they all lack effective BM structures (4, 24, 33, 42, 46), epidermal keratinocytes gradually lose their proliferative ability on the dermal scaffold. Therefore, more and more studies have focused on constructing dermal scaffolds with BM structures. Recent studies show that AM is an ideal source of scaffold materials for tissue engineering (28, 36). AM comes from the placenta with the properties of promoting epithelialization, alleviating inflammation, and inhibiting scarring. It has been widely used in the treatment of various corneal diseases, and AM tissue banks have been established in some countries (23, 28). AM can be considered an immune-privileged tissue with low immunogenicity (9, 15). Many studies reported that AM had good histocompatibility for xenografting/ allografting and did not induce significant immune rejection response (9, 15, 20, 22). Ma et al. (22) and Tsai et al. (40) used AM as the carrier to cultivate limbal stem cells for corneal reconstruction. Liang et al. used AAM to cultivate bone marrow mesenchymal stem cells for nerve repair (20). In the present study, we used AM to prepare a dermal scaffold with intact BM and good biostability. The results showed that it markedly increased the proliferating rate of EKs in vitro and promoted wound healing and dermal reconstruction after being grafted to full-thickness skin defects.
The process of AAM preparation directly affects its 3D structure and molecular compositions (36). Reported methods of AM decellularization include chemical treatment [such as ethylenediaminetetraacetic acid (EDTA) and sodium dodecyl sulfate (SDS)], enzymatic digestion (such as dispase and trypsin), and mechanical treatment (such as cell scraping and lyophillization) (14, 21, 37, 44). Chemical treatment and enzymatic digestion may cause lysis of the matrix proteins and the BM. Mechanical treatment does not affect the matrix structure significantly, but its decellularization effect is not good enough when used alone. Hopkinson et al. reported that thermophilic protease digestion could attenuate damage to the BM (10); however, it is difficult to apply it widely because of the low digestion rate and high cost. The repeated freeze– thaw method was initially used for decellularization of the embryoid body, meniscus, and nerves (7, 18, 27). In the present study, we, for the first time, used this method for AM decellularization treatment and compared it with the traditional dispase II digestion and cell scraping treatment. The results showed that repeated freeze–thaw cycles destroy cell membrane by forming crystals, causing cells to lyse, and the residual nuclei/DNA components are removed by additional DNase digestion effectively without significant influence on the matrix; thus, this method can effectively remove the cells in AM and maintain the integrity of the amniotic matrix, especially the BM structure. DispaseII digestion alone could not remove cells completely from the AM matrix, even when cell scraping treatment was used as an assistant method.
Proliferating EKs in normal skin are closely connected with the BM through hemidesmosomes, which can regulate the proliferation and differentiation of EKs and prevent cell apoptosis (1). It has been proved that both natural and artificial BM components can promote the proliferation of EKs (3, 13, 38, 41). The AAM we prepared in this study retained the intact BM structure and was able to provide a sound microenvironment for the adhesion and continuous proliferation of EKs. As this microenvironment is able to maintain cells in a continuous proliferating state, it raises the expansion rate of EKs in vitro, which is superior to the conventional culture method.
The disadvantage of AAM is that it is difficult to handle because it is very soft and has low mechanical strength and biostability. In addition, transplanted AM is likely to degrade quickly, especially in areas with severe inflammatory reaction (17, 23). We therefore used EDC to cross-link it. EDC is extensively used for strengthening mechanical strength and biostability of collagen, hyaluronic acid, acellular dermis, pericardium, and other biological tissues (31). It has been demonstrated that EDC cross-linked collagen materials can support the growth of human keratinocytes, fibroblasts, and mesenchymal stem cells (8, 25, 31). Our results showed that the extent of EDC cross-linking determines the mechanical strength and biostability of AAM: the higher the degree of EDC cross-linking, the higher the mechanical strength and the better the biostability. This is consistent with the results of previous studies (22, 31). We further demonstrated that EDC cross-linking associated the collagen and tropocollagen molecules of AAM with adjacent ones to form larger and more extensive fibrous bundles or fibrous cords, which determine the mechanical strength of AAM (29), thus raising its mechanical strength. However, the ductility of AAM also decreased at the same time.
Theoretically, EDC is converted to removable water-soluble urea during cross-linking and would not become part of the cross-linked material so that it is considered as a clean cross-linking method (16, 17). However, some studies reported that the cross-linked material was toxic when the concentration of EDC exceeded 10 mM (31). In our experiment, we did not find significant adverse effects of extracted solution prepared from different cross-linked AAMs on human EKs and fibroblasts (data not shown), but we found that the viability of EKs on the 30min-AAM and 6h-AAM decreased to some extent when they were cultured in a direct contact way, as the numbers of both dead and apoptotic cells increased significantly. However, there was no significant difference between the 5min-AAM and non-cross-linked AAM groups. Based on these findings, we supposed that residual EDC, urea, and intermediate reaction products such as O-acylisourea would be nonspecifically bounded on the surface of the cross-linked AAM and could not be removed completely. However, only when the degree of cross-linking is high enough (such as exceeding 82.28 ± 1.44% in the present experiment) would significant cytotoxicity appear.
The 5min-AAM in this study retained the BM structure of AM and its mechanical strength and biostability were also improved through appropriate EDC cross-linking. Further studies demonstrated good histocompatibility and low immunogenicity of the 5min-AAM, as no significant inflammatory or rejection response was observed after subcutaneous embedding, and only infiltration of a small number of inflammatory cells was observed. In addition, as the 5min-AAM was more resistant against collagenase and degraded gradually, it could be used as a dermal scaffold to construct composite skin substitutes. When grafted to full-thickness skin defects, the reticular fibrous structure of the 5min-AAM ensured good permeability of plasma, protein, and other nutrients in the early stage and provided a moist environment that is critical for rapid reepithelialization. The grafts survived well and fibroblasts entered the matrix and secreted collagens during the course of gradual degradation of the AAM. Vascularization of the scaffold and dermal reconstruction were significantly better than those of the control groups. Through interaction with the preexisting BM, the proportion of proliferating EKs at the BM increased significantly, forming a thicker and better stratified epidermis with a more complete BM structure. Four weeks after grafting, the BM formed in our model was continuous and ultrastructurally well-organized along the interface between the epidermis and dermis, as shown by immunolabeling for the classical BM marker laminin. The wound contraction was significantly improved in the EK-AAM groups.
Although the cross-linked AAM showed a lot of advantages in the cell culture and animal experiment, toxic and foreign body reactions cannot be completely excluded in clinical use of the cross-linked AAM, due to the limitations of the animal model used in this study and the difference in reactions to the cross-linked AAM between humans and animals; therefore, further observation is still needed in future studies.
Conclusion
In this study, we used a repeated freeze–thaw cycles and DNase digestion method to decellularize AM and then cross-linked it with EDC appropriately. The cross-linked AAM obtained has not only an intact BM structure but stronger mechanical strength and better biostability, thus promoting the adhesion and proliferation of human EKs. The grafted cross-linked AAM offers a biochemical and biophysical microenvironment required to modulate dermis formation by mimicking the function and structural characteristics of the native ECM. It can therefore be hopefully used as an ideal dermal scaffold and in material science and biological tissue engineering.
Footnotes
Acknowledgments
This project was supported by the National Natural Science Foundation of China (C30600646, 81071555, 30730091). The authors declare no conflict of interest.