
Abstract
Pluripotent stem cells represent an attractive cell source for regenerative medicine. However, the risk of teratoma formation after transplantation restricts their clinical application. Therefore, to adequately evaluate the potential risk of tumorigenicity after cell transplantation into human tissues, effective animal transplantation assays need to be developed. We performed a multiparameter (cell number, transplantation site, cell type, host) comparative analysis of the efficiency of tumor development after transplantation of mouse and human embryonic stem (ES) cells and their malignant counterparts, teratocarcinoma (EC) cells, into animal recipients and revealed several key correlations. We found that the efficiency of tumor growth was higher after intra-peritoneal than after subcutaneous transplantations of all cell lines studied. The minimal cell numbers sufficient for tumor growth in immunodeficient nude mice were 100-fold lower for intraperitoneal than for subcutaneous transplantations of mouse and human ES cells (103 vs. 105 and 104 vs. 106, respectively). Moreover, mouse ES and EC cells formed tumors in immunodeficient and immunocompetent mice more effectively than human ES and EC cells. After intraperitoneal transplantation of 103, 104, and 105 mouse ES cells, teratomas developed in 83%, 100%, and 100% of nude mice, whereas after human ES cell transplantation, teratomas developed in 0%, 17%, and 60%, respectively. In addition, malignant mouse and human EC cells initiated tumor growth after intraperitoneal transplantation significantly faster and more effectively than ES cells. Mouse and human ES cells formed different types of teratomas containing derivatives of three germ layers but different numbers of undifferentiated cells. ES cell-like sublines with differentiation potential similar to the parental cell line were recloned only from mouse, but not from human, ES cell teratomas. These findings provide new information about the possibility and efficiency of tumor growth after transplantation of pluripotent stem cells. This information allows one to predict and possibly prevent the possible risks of tumorigenicity that could arise from stem cell therapeutics.
Introduction
Pluripotent stem cell lines derived from the early embryos and experimentally converted from adult somatic cells have unlimited self-renewal potential and the ability of differentiation into multiple somatic cell types. Therefore, the nature of pluripotent stem cells makes them an ideal cell source for therapy of different diseases and recovery of injured tissues. However, one of the major challenges of pluripotent stem cell-based therapy is incomplete in vitro cell differentiation, which results in variable percentages of residual undifferentiated cells in transplants that may form teratomas in recipient tissues.
In the recent decade, numerous methods of in vitro differentiation of pluripotent stem cells into different cell types have been developed, and some of them have been tested in animal models of human diseases (2, 8, 22, 26, 43, 48, 50, 53). These studies have shown that, in most cases, the resulting cell populations are heterogeneous, and therefore, enrichment and selection of desirable cell types as well as elimination of the tumorigenic residual undifferentiated cells are required (3, 4, 43, 46). However, significant variations in the tumorigenic potential of transplanted cells were described (32, 43, 53). These contradictory outcomes may be due to the different in vitro differentiation protocols used and very short observation time during the experiments (commonly, 2–3 months), which may be insufficient to test tumorigenic potential. On the other hand, different transplantation techniques and tissue sites of transplantation, as well as genetic background and immunological state of recipient animals, also contribute to diverse developmental and tumorigenic potentials of the transplanted cells.
Tumorigenic potential variations may also be closely associated with the accumulation of genetic and epigenetic changes in the pluripotent stem cells arising during long-term cultivation in vitro (6, 15, 27, 33). Some of these may lead to cell death, impairment of cell functions, or enhancement of tumorigenicity (6, 15, 21, 47). Undifferentiated pluripotent stem cells or differentiating progenitor cells that acquired mutations in oncogenes and tumor suppressor genes and induced pluripotent stem cells derived using V-Myc myelocytomatosis viral oncogene homolog (C-myc) and Krüppel-like factor 4 (Klf4) transduction face a high risk of carcinogenesis (32). The oncogenic mutations were revealed in different pluripotent stem cell lines using genome-wide technologies for high-resolution analysis of genetic integrity (15, 27, 33). However, detection of rare mutations in single cells may be markedly limited, and therefore, the risk of carcinogenesis of transplanted cells still remains. In this context, to prevent all potential risks of tumorigenesis for prospective clinical application of pluripotent stem cell derivatives, the integrated technological platform for effective evaluation of stem cell-based therapeutic safety must include both large-scale genome monitoring of the genetic integrity of pluripotent stem cells and experimental transplantation assays. However, notwithstanding the extreme importance of these issues, there is as yet no standard procedure for experimental evaluation of the tumorigenic potential of pluripotent stem cell-based therapeutics, although clinical trials of human ES cell derivatives for therapies have already been approved by FDA and have begun.
Previous studies have shown that the growth of teratomas after transplantation of pluripotent stem cells into different animal models may depend on numerous factors (9, 11, 12, 24, 37, 44). However, the question on how to predict possible tumorigenicity of human embryonic stem (ES) cells after allogeneic/autological transplantations based on the outcomes of allogeneic/syngeneic transplantation of mouse ES cells and xenogeneic transplantation of human ES cells is still open. Moreover, it is difficult to compare the variable outcomes of transplantation studies with different experimental designs and techniques used in previously published works. Therefore, we performed a comparative multiparameter study of the growth and development of teratomas and teratocarcinomas after transplantation of mouse and human ES cells and their known malignant counterparts, mouse and human teratocarcinoma (EC) cells, maintained under similar conditions and transplanted using similar techniques. We analyzed several parameters simultaneously (cell, number, transplantation sites, normal and malignant cells, and hosts) and monitored tumor development within 50 weeks or until animal natural death. Thus, this is the first strict parallel analysis of tumor growth efficiency after transplantation of mouse and human ES cells as well as their genetically abnormal malignant analogs, EC cells.
Materials and Methods
Cell Culture
Mouse ES cell line R1 (derived from hybrid male blastocyst 129X1/SvJ and 129S1/SV-+p+Tyr-cKitlS1-J/+; 40,XY karyotype) was kindly provided by Prof. A. Nagy (Mount Sinai Hospital, Toronto, Canada), and human ES cell line ESM01 (46,XX karyotype) was kindly provided by Prof. G. P. Georgiev (Institute of Gene Biology, Russian Academy of Sciences, Moscow, Russia). Mouse and human teratocarcinoma cell lines F9 and PA-1 were obtained from Russian Cell Culture Collection (Institute of Cytology, Russian Academy of Sciences, St. Petersburg, Russia; http://www.rccc.cytspb.rssi.ru/). Mouse embryonic fibroblasts (MEFs) were derived from E13.5 fetuses of C57Bl/6 mice and used as feeder cells for routine ES cell maintenance.
Undifferentiated mouse and human ES cells were maintained on MEFs inactivated by mitomycin C (10 μg/ml; Sigma-Aldrich, St. Louis, MO, USA) in Dulbecco's modified eagle's medium (DMEM) containing 15% characterized fetal bovine serum (Hyclone, Logan, UT, USA) or knockout serum replacement (Gibco Invitrogen, Carlsbad, CA, USA), 2 mM l-glutamine, 0.1 mM nonessential amino acids (Hyclone), 0.1 mM β-mercaptoethanol, and 20 μg/ml gentamycin (Sigma-Aldrich). ES cell medium was supplemented with 10 ng/ml human basic fibroblast growth factor (bFGF; Gibco Invitrogen) for human ES cell maintenance and 10 ng/ml leukemia inhibitory factor (LIF, Sigma-Aldrich) for mouse ES cells cultivated under feeder-free conditions.
Karyotyping
Karyotype analysis was performed as described previously (30). For karyotyping, 30 metaphase plates of mouse and human ES cells were analyzed using VideoTesT-Karyo 3.1 software (Videotest, St. Petersburg, Russia).
Preparation of Cells for Transplantation
All experiments were performed using early and middle passage ES cells (ES R1 cells, passage 15–25; ESM01 cells, passage 35–45). The cells were prepared by dissociation of R1, F9, and PA-1 cell lines using 0.05% trypsin-EDTA solution (Hyclone). To prevent cell death, human ES cell colonies were manually cut into cell clusters and collected in four-well plates (Nalge Nunc International, Penfield, NY, USA) 1 h prior to transplantation. Finally, before subcutaneous transplantation, appropriate numbers of graft cells were concentrated in 50–70 μl of Hanks solution (Hyclone). For intraperitoneal (under the kidney capsule) and subcutaneous transplantations of small cell numbers (10–10,000), cells were placed in 10- to 15-μl drops of Hanks solution and then transplanted into animals using plastic capillaries (Cook Flexipet Pipettes, Cook Medical, Bloomington, IN, USA) or pulled glass micropipettes of 140- and 300-μm inner diameter.
Animal Experiments
In order to study the development of teratomas and teratocarcinomas, we used as recipients immunodeficient nude mice (Nu/Nu) and immunocompetent C57Bl/6 mice from the Animal Breeding Facility-Branch “Pushchino” of Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences (stock line was obtained from Charles River Laboratories, Inc., Wilmington, MA, USA). Mice were kept under pathogen-free conditions. All animal study protocols were approved by the Institutional Bioethical Committee. For experiments, recipient mice at the age of 3–4 months were used. Before the subcutaneous and intraperitoneal (under the kidney capsule) cell transplantation procedures, the animals were anesthetized by an intraperitoneal injection of 100 mg/kg ketamine (MEZ, Russia) and 10 mg/kg xylazine (Rometar, Spofa, Czech Republic). In subcutaneous transplantation experiments, mouse and human ES and EC cells were injected under the skin of the back area of nude and C57Bl/6 mice using 1-ml syringes with 27-gauge needles (Becton Dickinson, Franklin Lakes, NJ, USA) or pulled glass micropipettes (100- to 300-μm inner diameter).
The procedure of ES and EC cell transplantation under the kidney capsule was carried out under a surgical microscope (Carl Zeiss, Jena, Germany). Lumbar incisions in the skin and body wall were made to get access to the kidney. The renal capsule of kidney apex was held with forceps and punctured with a needle, avoiding organ injury. The cells have been injected under the kidney capsule using a plastic capillary (Cook Medical) connected to a CellTram Vario Eppendorf Microinjector (Eppendorf, Hamburg, Germany). At the end of experiments, the animals were sacrificed by cervical dislocation or by injecting a lethal dose of intravenous barbiturates, and autopsies of all mice were performed. Developed tumors were removed and weighted. Statistical analyses of tumor weight were performed using one-way ANOVA and Student–Newman–Keuls test. Differences were considered significant at p < 0.05.
Recloning of Mouse and Human ES and EC Cells From Developed Tumors
To investigate the differentiation and tumorigenic potentials of the residual undifferentiated cells in teratomas and teratocarcinomas, tumor tissue samples were dissected into small pieces and dissociated by treatment with 0.25% trypsin-EDTA solution (HyClone). The resulting cell suspensions were transferred in six-well plates (Nalge Nunc) in ES cell medium for the following cultivation in vitro within 5–7 days. Mouse ES cell colonies were picked from mixed cultures and transferred to gelatin-coated plates (Sigma) in ES cell medium supplemented with LIF for following expansion of the clones.
Histochemical, Immunofluorescence, and Histological Analyses
For immunofluorescence analysis and alkaline phosphatase activity assays, ES and EC cells were fixed by 2% paraformaldehyde in phosphate-buffered solution, pH 7.0 (Sigma-Aldrich). The activity of alkaline phosphatase in the cells was detected after incubation in a solution containing 10 ml of 0.002 M Tris-HCl buffer (pH 8.7), 1 mg of naphtol-AS-B1-phosphate, and 5 mg of Fast Red dye Texas Red (Sigma-Aldrich) at 37°C for 1 h. Immunofluorescence staining of ES and EC cell colonies was performed as described previously (30) using antibody against octamer-binding transcription factor 3/4 (OCT3/4; 1:150, rabbit IgG, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
For histological analysis, all developed tumors were isolated during autopsy, and tumor samples were fixed in Bouin's solution (Sigma-Aldrich). Tumor samples were dehydrated according to the standard method and embedded in paraffin for sectioning. Histological sections were stained by hematoxylin and eosin (Sigma-Aldrich) and examined under a Leica DMRXA2 microscope (Leica Microsystems, Wetzlar, Germany).
RNA Isolation and RT-PCR Analysis
Total RNAs were extracted from mouse and human ES and EC cells and from teratoma and teratocarcinoma samples using TRIzol reagent (Invitrogen) according to the manufacturer's recommendations. Each sample was treated with RNase-free DNase (TurboDnase, Ambion, Life Technologies, Grand Island, NY, USA) to avoid DNA contamination. For cDNA libraries synthesis, 1 μg of total RNA from each sample was reverse-transcribed using RevertAid M-MuLV revertase and random hexamer oligonucleotide primers (Fermentas UAB, Vilnius, Lithuania). The PCR reaction mixtures were prepared according to the manufacturer's protocol (protocol for Taq polymerase, Fermentas). The probes were denatured at 94°C for 5 min and cycled at 94°C for 45 s, 56–58°C for 45 s, and 72°C for 60 s, followed by a final extension at 72°C for 5 min after completion of 35 cycles. For detection of mouse gene expression, the following primer sequences and their expected products were used: Oct4 (f) 5′-tggagactttgcagcctgag-3′, (r) 5′-catactcttctcgttgggatta-3′ (621 bp); Nanog (f) 5′-caagcggtggcagaaaaac-3′, (r) 5′-tggataagagcacccgactg-3′ (702 bp); alpha fetoprotein (Afp) (f) 5′-agctcagcgaggagaaatgg-3′, (r) 5′-caaaaggcccgagaaatctg-3′ (350 bp); hypoxanthine phosphoribosyltransferase 1 (Hprt) (f) 5′-gctggtgaaaaggacctct-3′, (r) 5′-cacaggactagaacacctgc-3′ (249 bp). The following primers were used for studying human gene expression: OCT4 (f) 5′-gggtggaggaagctgacaac-3′, (r) 5′-gcatagtcgctgcttgatcg-3′ (259 bp); NANOG (f) 5′-ctgcgtcacaccattgctattc-3′, (r) 5′-tgcctcacacggagactgtc-3′ (368 bp); AFP (f) 5′-gcggcctcttccagaaacta-3′, (r) 5′-tttcatccaccaccaagctg-3′ (312 bp); ribosomal protein L19 (RPL-19) (f) 5′-agggtacagccaatgcccga-3′, (r) 5′-ccttggataaagtcttgatgatc-3′ (326 bp).
Results
Developmental Kinetics of Mouse and Human ES and EC Cells After Transplantation Into Immunodeficient Nude Mice
In order to determine the developmental kinetics of the tumors formed by mouse and human pluripotent and teratocarcinoma cells, 106 undifferentiated cells of each cell line were injected subcutaneously or intraperitoneally into nude mice, and tumor development was monitored within 4–12 weeks. During this time, teratomas and teratocarcinomas developed in all mice (100%), but the growth dynamics of the transplanted cells differed (Fig. 1 and Table 1). It can be seen that equal numbers of mouse ES and EC cells formed visible tumors in recipients within a shorter period of time than human cells. Mouse and human teratocarcinomas grew more rapidly and intensively than the teratomas formed by appropriate ES cells. In most cases, more intensive tumor growth was observed after intraperitoneal transplantation. However, we never detected tumor growth, even for teratocarcinoma cells, outside the transplantation sites. Based on histological analysis, the optimal experimental times for studying tumor development were determined for each cell line (Table 1).
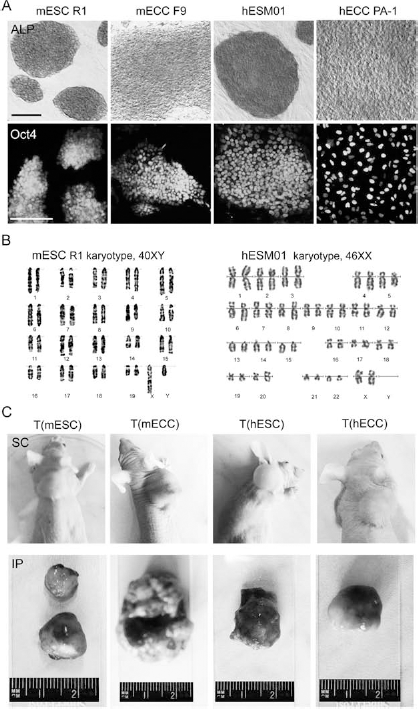
Mouse and human ES and EC cell growth in vitro and after transplantation into immunodeficient mice. (A) Undifferentiated mouse and human embryonic stem (ES) and teratocarcinoma (EC) cells express high activities of alkaline phosphatase (ALP) and key pluripotency transcriptional factor octamer binding transcription factor 3/4 (Oct3/4). Scale bar: 100 μm. (B) Mouse ES R1 and human ESM01 cell lines display normal karyotypes. (C) Macroscopically, teratomas and teratocarcinomas developed after subcutaneous and intraperitoneal (under the kidney capsule) transplantations into nude mice. mESC R1, mouse ES R1 cells; mECC F9, mouse EC F9 cells; hESM01, human ESM01 cells; hECC PA-1, human EC PA-1 cells; T(mESC), T(mECC), T(hESC), T(hECC), tumors developed after subcutaneous (SC) and intraperitoneal (IP) transplantations of mouse and human ES and EC cells, respectively.
Efficiency of Tumor Development After Transplantation of Mouse and Human ES and EC Cells Into Immunodeficient Nude Mice

The percentage and number of animals are indicated in which tumors were found in the transplantation sites during autopsy. ESC, embryonic stem cell; ECC, teratocarcinoma cell; m, mouse; h, human; SC, subcutaneous transplantation; IP, intraperitoneal transplantation (under the kidney capsule).
p < 0.05, weights of tumors developed after subcutaneous and intraperitoneal transplantations significantly differed from each other for each cell line.
Minimal Cell Numbers Inducing Tumor Development After Intraperitoneal and Subcutaneous Transplantations of Mouse and Human ES and EC Cells Into Immunodeficient Nude Mice
To investigate the minimal cell numbers inducing tumor development after transplantation of mouse and human ES and EC cells into different transplantation sites, the undifferentiated mouse ES and EC cells (from 10 to 105 cells) were transplanted under the kidney capsule or under the skin of nude mice. The observation times of tumor growth in mice that received small cell numbers were extended to 1 year. We found that the minimal cell numbers initiating tumor development intraperitoneally were 100 and 1,000 cells for mouse teratocarcinoma F9 line and mouse ES R1 line, respectively. However, for subcutaneous transplantation, the minimal cell numbers were similar for both cell lines at 105 cells (Table 2).
Tumor Development After Intraperitoneal and Subcutaneous Transplantations of Different Cell Numbers of Mouse ES and EC Cells Into Immunodeficient Nude Mice
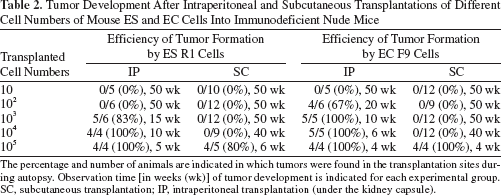
The percentage and number of animals are indicated in which tumors were found in the transplantation sites during autopsy. Observation time [in weeks (wk)] of tumor development is indicated for each experimental group. SC, subcutaneous transplantation; I P, intraperitoneal transplantation (under the kidney capsule).
The results of experiments with human ES and EC cells have shown that the minimal cell numbers that can induce human teratoma and teratocarcinoma growth in xenogeneic environment were 10-fold higher than for mouse cells (Table 3). For intraperitoneal transplantation of human ES and EC cells, they were 104 and 103 cells, respectively, although the efficiency of tumor development was about 20% for ES cells and 60% for EC cells. Like the mouse cell lines, for both human cell lines the minimal cell numbers initiating tumor growth after subcutaneous transplantation were similar for 0.5–1×106 cells; that is, significantly higher than for intraperitoneal transplantation. Moreover, the tumor size and histological contents of teratomas depended on graft cell numbers and time of development. We observed that human ES cells transplanted in small numbers grew slowly and formed very small tumors, which did not contain cell derivatives of all three germ layers.
Tumor Formation After Intraperitoneal and Subcutaneous Transplantations of Different Numbers of Human ES and EC Cells Into Immunodeficient Nude Mice

The percentage and number of animals are indicated in which tumors were found in the transplantation sites during autopsy. Observation time [in weeks (wk)] of tumor development is indicated for each experimental group. SC, subcutaneous transplantation; IP, intraperitoneal transplantation (under the kidney capsule).
Minimal ES and EC Cell Numbers Inducing Tumor Development in Immunocompetent Mice
At the next step, we compared the efficiency of tumor development in hosts with different immune states, immunodeficient and immunocompetent allogeneic mice. In our experiments, the immunocompetent allogeneic C57Bl/6 mice did not develop tumors after both intraperitoneal and subcutaneous transplantations of 1–3×106 human ES and EC cells within 1 year (Table 4). On the contrary, mouse ES and EC cells formed teratomas and teratocarcinomas in most of allogeneic C57Bl/6 mice even without immunosuppressive treatment, but cell numbers initiating tumor growth were significantly higher than for nude mice (Table 4 and Fig. 2). Most allogeneic C57Bl/6 mice that received 3×106 mouse ES cells subcutaneously or 3×104 cells intraperitoneally developed teratomas. Similarly, minima of 106 and 103 mouse EC cells formed teratocarcinomas in subcutaneous or intraperitoneal sites, respectively. Compared with the immunodeficient mice, these minimal cell numbers were approximately 10- to 30-fold higher. Thus, our study demonstrates that the immune system of adult immunocompetent C57Bl/6 mice effectively prevents tumor growth after injection of large numbers of xenogeneic human ES and even EC cells but does not reject large numbers of mouse ES and EC cells.

Teratomas and teratocarcinomas following minimal effective number of cells transplanted into C57Bl/6 and nude mice. (A) Teratomas [T(mESC)] and teratocarcinomas [T(mECC)] developed after subcutaneous (SC) and intraperitoneal (IP) transplantations of minimal effective numbers of mouse ES and EC cells into C57Bl/6 and nude mice. All tumors developed within 4 weeks. (B) Immunological response and destruction of teratoma formed after transplantation into C57Bl/6. (C) The section through the kidney of a nude mouse transplanted with 100 mouse ES cells. No teratoma or other neoplasms were found at 50 weeks after transplantation. Scale bar: 200 μm. (D) Examination of human OCT4, ribosomal protein L19 (RPL19), and mouse hypoxanthine phosphoribosyltransferase (Hprt) expression in kidneys of nude mice that did not develop tumors after transplantation of human ES and EC cells. K1(hESC), K2(hESC): transplantation of 1,000 hES cells; K3(hECC), K4(hECC): transplantation of 100 hEC cells; T(hESC), T(hECC): tumors developed after transplantation of human ES and EC cells.
Tumor Development After Transplantation of Mouse and Human ES and EC Cells Into Immunocompetent C57Bl/6 Mice

The percentage and number of animals are indicated in which tumors were found in the transplantation sites during autopsy. Observation time [in weeks (wk)] of tumor development is indicated for each experimental group. SC, subcutaneous transplantation; IP, intraperitoneal transplantation (under the kidney capsule).
Differentiation Patterns of Mouse and Human ES Cells in Teratomas
Histological analysis of the teratomas that developed after intraperitoneal and subcutaneous transplantations of the mouse and human ES cells has shown that the cellular contents of tumors did not differ markedly. No differences in histological contents of the teratomas formed by mouse ES cells after transplantation into immunodeficient or immunocompetent mice were found, but spacious areas with lymphocyte infiltration were present in the teratomas developed in immunocompetent allogeneic mice (Fig. 2B). We monitored neoplasm formation under the skin in mice lacking teratomas under a surgical microscope during autopsy, but no surviving transplanted cells were detected. No benign or malignant neoplasms were found in the kidneys or other organs of peritoneal cavity of nude mice that received low cell numbers at 50 weeks after transplantation (Fig. 2C). PCR analysis of OCT4 or RPL19 expression was performed to examine some kidneys after hES and hEC cell transplantations, but the presence of transplanted human cells was not confirmed (Fig. 2D).
All teratomas studied contained different cell derivatives of three germ layers. Teratomas formed by both mouse and human ES cells contained numerous tubular structures with different types of epithelium of ectodermal (keratinized, exfoliated, neuroectodermal) or endodermal (secretory, ciliated, cuboid) origin (Fig. 3). However, mesodermal derivatives—striated and smooth muscles, connective and adipose tissues, cartilage and bone—represented the main bulk of teratomas (Fig. 3). Note that the teratomas formed by small numbers of mouse ES cells contained representatives of three germ layers, while in the teratomas formed by small human ES cell numbers, the derivatives of not all germ layers were detected.

Histological contents of teratomas and teratocarcinomas formed after transplantation of mouse and human ES and EC cells into nude mice. (A) Sections through teratomas and teratocarcinomas formed by mouse ES and EC cells. Teratomas contain derivatives of three germ layers. (B) Teratomas and teratocarcinomas formed by human ES and EC cells. Cell derivatives of ectoderm, mesoderm, and endoderm in teratomas. (C) Expression of gene markers of undifferentiated (Oct4, Nanog) and differentiated [alpha fetoprotein (Afp)] pluripotent stem cells in teratomas and teratocarcinomas formed by mouse ES and EC cells. (D) Expression of OCT4, NANOG, and AFP in teratomas and teratocarcinomas formed by human ES and EC cells. For designations, see Figure 1.
In the teratomas formed by mouse and human ES cells, the clusters of low differentiated cells remained and were integrated between differentiated tissues and structures. Moreover, the expression of transcriptional factors Oct4 and Nanog, which is characteristic of undifferentiated pluripotent stem cells, was detected in most teratomas formed by mouse ES cells. However, as opposed to mouse ES cells, OCT4 and NANOG expression in human teratomas was significantly lower than that in the mouse teratomas, suggesting a markedly smaller number of undifferentiated cells in human ES cell teratomas (Fig. 3).
Recloning of ES Cell Sublines From Developed Teratomas
In order to determine the differentiation potential of residual undifferentiated cells in teratomas, tumor samples were dissociated and cell suspensions were cultivated in vitro. After 3–5 days of cultivation, residual undifferentiated cells from the mouse ES cell teratomas formed ES cell-like colonies (Fig. 4). Numerous mouse ES cell sublines (44 sublines) were recloned from the primary teratoma cell cultures. All these sublines had characteristics and in vitro differentiation potential identical to the parental ES R1 cells. Three sublines were tested for the formation of secondary tumors. The secondary teratoma growth dynamics were similar to those of primary teratomas. All developed secondary teratomas contained cell derivatives of three germ layers as well as undifferentiated cells expressing Oct4 (Fig. 4). Mouse and human teratocarcinoma sublines were also recloned from developed tumors. They displayed specific characteristics of the parental cell lines.

Studies of growth and developmental potentials of three (R1.1, R 1.2, R 1.3) of the 44 sublines recloned from teratomas formed by mouse ES R1 cells after transplantation of mouse ES R1 cells into nude mice. The ES cell sublines tested and parental ES R1 cell line demonstrated similarity of their in vitro and in vivo differentiation characteristics. Undifferentiated R1.1, R 1.2, and R 1.3 cells maintained in vitro expressed ALP, Oct4, and Nanog and formed secondary teratomas after transplantation into nude mice. Developed secondary teratomas contained cell derivatives of three germ layers and Oct4- and Nanog-expressing cells. Scale bar: 100 μm.
However, we could not reclone human ES cell sublines from human teratomas using the same methods. This could be due to a very low survival ratio and clonogenicity of human ES cells after dissociation. Furthermore, it is well known that human ES cells have greater propensity to spontaneous differentiation in vitro than mouse ES cells, and this may be true for in vivo differentiation as well. It cannot be ruled out that the growth and differentiation potentials of the human ES cells transplanted into adult xenogeneic tissues differ from those of mouse ES cells transplanted into allogeneic hosts.
Discussion
Previous studies have shown that teratoma formation efficiency varied for different transplantation sites, especially under the skin, even after transplantation of large numbers of undifferentiated ES cells (9, 20, 29, 37, 43). At the same time, 100% efficiency of teratoma formation was observed after transplantation of 1 × 106 undifferentiated mouse ES cells subcutaneously in immunodeficient or syngeneic mice, in contrast to human cells (11). It was unclear whether this is inherent in human pluripotent stem cells transplanted into xenogeneic recipients or is also characteristic of all pluripotent stem cells. Therefore, we reinvestigated site-dependent tumor growth after transplantation of mouse and human ES and EC cells intraperitoneally and subcutaneously into immunodeficient nude mice. In our experiments, 1 × 106 mouse and human ES and EC cells transplanted intraperitoneally or subcutaneously resulted in teratoma development in all animals (100%), but tumor growth rates were more intense in the peritoneal cavity than under the skin for all cell lines studied. However, the most striking differences in tumor formation efficiency were found after transplantation of small cell numbers (Tables 2 and 3). The minimal cell numbers inducing tumor development after intraperitoneal transplantation were markedly less than for subcutaneous transplantation for all cell lines, although the minimal cell numbers inducing tumor formation under the skin were similar for ES and EC cells of each species. Taken together, our data indicate that the tumor growth efficiency considerably depends on the tissue site of transplantation for both mouse and human ES cells, and even EC cells. The site-dependent differences in mouse and human teratoma development may be due to several causes: firstly, the viability, growth, and differentiation rates of the transplanted cells; secondly, the immunological responses of the host immune system that may differ in different tissue sites even of immunodeficient mouse strains; and thirdly, the trophic conditions and vascularization into the transplantation sites (52). We observed that different techniques and depths of subcutaneous cell injections might also affect cell graft rejection. Thus, subcutaneous transplantation, widely used as a simple technique, may cause contrary and false-negative outcomes and, therefore, cannot be recommended as a reproducible test of pluripotent stem cell safety.
To determine safe borders for pluripotent stem cell derivative transplantations, it is necessary to know the minimal numbers of undifferentiated mouse and human ES cells sufficient for teratoma development in different tissue sites. Our comparative examinations of the efficiency of teratoma development after subcutaneous and intraperitoneal transplantations of different numbers of undifferentiated mouse and human ES cells showed that the minimal cell numbers sufficient for tumor development after transplantation into immunodeficient mice were 10-fold lower for mouse ES cells than for human ES cells in both transplantation sites. As mentioned above, these cell numbers were site dependent for both cell lines. Moreover, the minimal numbers of mouse and human EC cells inducing teratocarcinoma growth were at least 10-fold lower than for ES cell teratomas.
Similar critical cell numbers were determined for human ES cells transplanted into the myocardium and skeletal muscle of severe combined immunodeficient (SCID)/beige mice (105 and 104 cells, respectively) (25, 29) and mouse ES cells transplanted into the myocardium and brain (500–1,000 cells) (7, 18). In other studies, effective teratoma development was observed after transplantation of 105 undifferentiated human and cynomolgus monkey ES cells under both the testicular capsule and skin (23, 43) and 0.5×105 mouse ES cells transplanted into the heart (36). However, even 245 undifferentiated human ES cells mixed with 106 human fibroblasts and transplanted into the skeletal muscle of SCID mice were capable of developing teratomas within 12 weeks (20). Similarly, 2–20 mouse ES cells transplanted under the kidney capsule or subcutaneously with Matrigel resulted in 100% efficiency of tumor growth in both sites (28). Note that the Matrigel use may mask the bona fide tumor growth efficiency because it significantly enhances growth of any cells.
We found that the minimal cell numbers for initiating teratomas after transplantation in both transplantation sites of immunodeficient mice were lower for mouse ES cells than for human ES cells, but it cannot be ruled out that it would be similar in the case of autologous transplantation of human ES cells. In addition, the possible mutations leading to cancer transformation of pluripotent stem cells, like in their malignant counterparts, EC cells, may enhance the probability and efficiency of the tumor growth after transplantation because lower cell number can initiate tumors. Hence, the safety borders for pluripotent stem cell therapy are significantly reduced in these cases.
Mouse and human pluripotent stem cells transplanted in animal hosts differentiate into various somatic cell types in teratomas independently of transplantation site. However, the residual undifferentiated cells are retained even in well-developed teratomas. Recloned mouse ES cell sublines from teratomas display growth and differentiation potentials identical to those of the parental ES cell line. Unlike mouse teratomas, human ES cell teratoma dissociated and cultivated in vitro did not give rise to ES cell-like colonies. As mentioned above, human ES cells have low viability and clonogenicity after single-cell dissociation and are more prone to spontaneous differentiation than mouse ES cells. Therefore, we suggest that xenogeneic host tissues may not represent the optimal microenvironment for intense undifferentiated human ES cell growth and, like inappropriate in vitro culture systems, encourage their tendency to differentiate. On the other hand, it cannot be ruled out that residual undifferentiated cells may represent transformed cancer cells. Several aneuploid human ES cell lines have been found to grow more intensively in vitro and in teratomas, which contained more undifferentiated cells (1, 49). However, our study of primary and secondary tumors formed by mouse ES and EC cells as well as human EC cells showed no differences in their growth and differentiation potentials. We never observed tumors formed by ES cells that were predominately composed of undifferentiated cells as seen with teratocarcinoma tumors. Therefore, we suggest that mouse and human ES cells form basically different types of teratomas in immunodeficient mice, and despite the presence of undifferentiated cells in mouse ES cell teratomas, recloned ES cells are not EC-like cells with restricted differentiation potentials.
It is well known that human ES cells are not equivalent to mouse ES cells and represent later and more slowly dividing embryonic cell types that are congruous to epiblast cells (primed state) but not the inner cell mass cells as mouse ES cells (ground state) (19, 35). We believe that the similarity of human ES cells and mouse epiblast stem cells also suggests their predominant differentiation into somatic rather than germ cells. Conversely, mouse ES cells as genuine pluripotent cells can form teratomas consisting of somatic cells together with undifferentiated ES-like cells that may be considered as the earliest precursors of primordial germ cells. These cells can be converted easily into pluripotent stem cells in vitro, like embryonic germ cells from embryonic gonads (13). Moreover, primordial germ cells of different developmental stages have been shown to form teratomas, unlike the embryonic somatic cells of the same stages (13, 17, 34, 42). Undoubtedly, this hypothesis needs to be tested in further studies.
The efficiency of tumor growth after transplantation of pluripotent stem cells significantly depends on the recipient animal model used. We found similar trends of higher tumorigenicity of mouse ES and EC cells than human ES and EC cells after transplantations in both immunocompetent and immunodeficient mice. The immunocompetent C57Bl/6 mice did not develop teratomas after transplantation of human ES and EC cells, while mouse ES and EC cells formed tumors after intraperitoneal or subcutaneous transplantations into allogeneic immunocompetent C57Bl/6 mice, but the minimal cell numbers inducing tumor growth were significantly higher than for the immunodeficient nude mice. Moreover, tumor growth efficiency was also site dependent for immunocompetent mice (Table 4). Our data demonstrate that small numbers of mouse ES and EC cells were effectively rejected after transplantation in allogeneic immunocompetent recipients, while large cell numbers were capable of tumor formation, probably due to their immunosuppressive effect and high self-renewal rate. However, we observed intensive immunological response in teratomas and teratocarcinomas developed in immunocompetent mice. The absence of tumor growth after transplantation of small cell numbers into immunocompetent and immunodeficient mice may be due to the effective early response of different components of the immune system in the small cell transplants (10–12) and the slow increase of transplanted cell populations. On the other hand, the scattered and poorly attached cells transplanted may undergo apoptosis in the absence of required supporting stromal cells and extracellular matrix components. Therefore, a coinjection of Matrigel or fibroblasts with human and mouse ES cells may improve cell viability and enhance the tumor growth under the skin (20, 28, 37).
Previous studies demonstrated that the mouse, monkey, and human pluripotent stem cells developed teratomas after transplantation into different immunodeficient [SCID/ beige, nonobese diabetic/severe combined immunodeficient (NOD/SCID), SCID, recombination activating gene 1 knockout (RAG1–/–), Nude] or syngeneic mouse strains (5, 9, 11, 12, 14, 20, 23, 38, 43, 45), whereas immunocompetent allogeneic mice or xenogeneic recipients failed to develop teratomas after transplantation of the ES cells without immunosuppression (11, 12, 16, 39, 40). Nevertheless, teratomas were found in allogeneic immunocompetent mice after intramyocardial, intrahepatic, and subcutaneous transplantations of large numbers (1–20×106 cells) of mouse ES cells (24, 31, 36, 51). At the same time, the outcomes of xenogeneic transplantations of the mouse and human ES cells into immunodeficient or immunocompetent recipients are significantly variable and hardly predictable. Human ES cell-derived neurospheres transplanted into the injured spinal cords of immunodeficient HsdHan:RNU-Foxn1rnu rats gave rise to teratomas, unlike the transplantation into the testis of SCID mice (43). Similarly, hematopoietic precursor cells differentiated in vitro from cynomolgus monkey ES cells and transplanted into NOD/SCID mice or into the liver of sheep fetuses formed teratomas at a very low frequency, whereas tumors rapidly developed after allogeneic transplantation into the fetal liver of cynomolgus monkeys (39, 40). Undifferentiated cynomolgus monkey ES cells transplanted into the fetal sheep liver could form teratomas in the fetus at 43–49 gestational days, but not after 50 gestational days (45). In addition, most teratomas formed by human ES cells after transplantation in adult immunodeficient animals represent benign differentiated tumors, but after transplantation of human ES cells into human fetal tissues engrafted previously in SCID mice, aggressive growth of primitive undifferentiated tumors was detected (41). Thus, the present and previous findings show that the efficiency of tumor growth after transplantation into allogeneic and xenogeneic animals depends on the crucial balance between host regulatory immune cell response and the adaptive capability and growth rate of transplanted cells.
In summary, our comparative investigation of the growth and differentiation potentials of mouse and human ES and EC cells after transplantation in mice showed that the efficiency of tumor development depends on the transplantation site and transplanted cell number for all cell lines studied. Therefore, to predict the possible tumorigenicity of human ES cell derivatives after allogeneic/autologic transplantation, several transplantation sites must be tested in each pluripotent stem cell-based therapeutic safety study independently of therapy model studies. Subcutaneous transplantation cannot be recommended as a reproducible and reliable test of tumorigenicity because it displays the lowest efficiency of tumor growth. For effective evaluation of safety of pluripotent stem cell-derived therapeutics using animal transplantation assays, the outcomes of allogeneic transplantations of mouse ES cells need to be taken into account because xenogeneic animal transplantation assays demonstrate understated risks of tumorigenicity. Hence, for safety testing, transplanted cell numbers must be high (at least 107–108 cells) because the present study has shown that the efficiency of tumor growth in immunodeficient nude mice was lower after transplantation of human ES cells than after transplantation of mouse ES cells. Moreover, according to our findings, the minimal numbers of mouse and human EC cells sufficient for inducing teratocarcinoma growth in immunodeficient mice were 10-fold lower compared to normal mouse and human ES cells; this indicates a high probability of tumor growth in the case of cancerous transformation of pluripotent stem cells like EC cells. Taking into account that none of the currently used animal models can be recognized as the most effective for human pluripotent stem cell safety studies, new humanized animal models are necessary for the development of stem cell therapeutic safety evaluation technology.
Footnotes
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (grants 05-04-49185, 11-04-00379) and the Russian Academy of Sciences Program “Molecular and Cell Biology.” The authors thank A. Hovsepian and G. Telegin (Animal Breeding Facility-Branch “Pushchino” of Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences) for their assistance with animal care, and N. Krasnikova for assistance with RT-PCR. They also thank Dr. Sergei Vassetsky for critical reading of the manuscript. The authors declare no conflict of interest.