
Abstract
Induced pluripotent stem (iPS) cells may provide cures for various neurological diseases. However, undifferentiated iPS cells have high tumorigenicity, and evaluation of the cells fates, especially in pathologic condition model, is needed. In this study, we demonstrated the effect of ischemic condition to undifferentiated iPS cells fates in a mouse model of transient middle cerebral artery occlusion (MCAO). Undifferentiated iPS cells were characterized with immunofluorescent staining. The iPS cells (5 × 105) were injected into ipsilateral striatum and cortex after 24 h of MCAO. Histological analysis was performed from 3 to 28 days after cell transplantation. iPS cells in ischemic brain formed teratoma with higher probability (p < 0.05) and larger volume (p < 0.01) compared with those in intact brain. Among the four transcriptional factors to produce iPS cells, c-Myc, Oct3/4, and Sox2 strongly expressed in iPS-derived tumors in ischemic brain (p < 0.01). Additionally, expression of matrix metalloproteinase-9 (MMP-9) and phosphorylated vascular endothelial growth factor receptor2 (phospho-VEGFR2) were significantly increased in iPS-derived tumors in the ischemic brain (p < 0.05). These results suggest that the transcriptional factors might increase expression of MMP-9 and activate VEGFR2, promoting teratoma formation in the ischemic brain. We strongly propose that the safety of iPS cells should be evaluated not only in normal condition, but also in a pathologic, disease model.
Introduction
Induced pluripotent stem (iPS) cells are somatic cells that have been reprogrammed to enter a pluripotent state. Yamanaka et al. first established a murine iPS cell line by introducing four transcriptional factors, c-Myc, Sox2, Oct3/4, and Klf4, into murine fibroblasts (30). Human iPS cell lines have also been established with similar methods (23,31). iPS cells can be produced from each patient's skin, and retain high replication competence and pluripotency to differentiate various kinds of cells, indicating that iPS cells can be a promising cell source for cell transplantation therapy (2,13,25,33).
Strokes are a major cause of death and a reduction in the quality of life caused by a stroke is a serious problem for which effective therapy is not yet available. Stem cell transplantation therapy is regard as a promising strategy to supply newly born neurons repairing disrupted neuronal networks of the postischemic brain (1,4,5,17). However, some researchers have reported that mouse or primate embryonic stem cells generated teratomas when transplanted into immunodeficient mice (7,11). Germline-competent chimera mice with iPS cells also develop tumors (20). A recent study showed that a secondary neurosphere from iPS cells formed a teratoma in mouse brains at a constant rate (18). They reported that the persistence of undifferentiated iPS cells, which remain undifferentiated and in a pluriopotent state even after a differentiation assay, formed a teratoma. This high tumorigenicity interferes with clinical applications, but the fate of undifferentiated iPS cells transplanted in a pathologic condition model such as ischemia remains unknown.
Here we transplanted undifferentiated murine iPS cells in normal or ischemic striatum/cortex of the mouse brain, and observed their fate. We found that iPS cells transplanted in ischemic brain formed a teratoma with higher probability, and with larger volume compared with those in the normal brain.
Materials and Methods
Animals and Experimental Groups
Adult (20–25 g, 8–10 weeks old) male C57BL/6N mice were used in this study. All experimental procedures were approved by the Animal Committee of the Okayama University Graduate School of Medicine. We studied six experimental groups: Sham + PBS group (n = 6), MCAO + PBS group (n = 8), Sham + iPS group (n = 8), MACO + iPS group (n = 11), Sham + virus-free iPS group (n = 3), and MACO + virus-free iPS group (n = 4). Each experimental group received intracerebral implantation of PBS or murine iPS cells at 24 h after sham operation or MCAO, as described below.
Focal Cerebral Ischemia
During surgery, the mice were anesthetized with a nitrous oxide/oxygen/isoflurane mixture (69%/30%/1%) administered through an inhalation mask. MCAO was induced by the intraluminal filament technique (32). In brief, the right carotid bifurcation was exposed, and the external carotid artery was coagulated distal to the bifurcation. A silicone-coated 8–0 filament was then inserted through the stump of the external cerebral artery and gently advanced (9.0–10.0 mm) to occlude the middle cerebral artery. After 30-min occlusion, the filament was gently withdrawn, and the incision was closed. Rectal temperature was maintained at 37.0°C by placing the animals on a heating bed (model BMT-100; Bio Research Center). A laser Doppler flowmeter probe (MBF3D, Moor Instruments Ltd) was attached to the surface of the ipsilateral cortex to monitor regional cerebral blood flow.
Preparation and Transplantation of iPS Cells
Murine iPS cell lines (iPS-MEF-Ng-20D-17, iPS-MEF-Ng-492B-4) were provided by RIKEN BRC through the National Bio-Resource Project of MEXT, Japan. The iPS cells were maintained on a feeder layer of mitomycin C-treated mouse embryonic fibroblasts (Millipore) in Dulbecco's modified Eagle medium containing 15% fetal bovine serum, 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol, and 1000 U/ml mouse leukemia-inhibiting factor (29).
For transplantation, the iPS cells were collected, suspended, and incubated with fresh medium on 0.1% gelatin-coated dishes for 30 min to remove contaminated mouse embryonic fibroblasts. The iPS cells were collected and labeled with Qtracker 565 cell labeling kit (Invitrogen) according to the manufacturer's specifications. Labeled cells were washed three times with PBS and pelleted by centrifugation. The pellets were resuspended in 15 μl PBS and placed on ice. Two microliters of the cell suspension (5 × 105 cells) or 2 μl of PBS was stereotaxically injected into the ipsilateral striatum and cortex [anterior, lateral, depth (in mm): −0.5, 2.5, 1.5–2.5] (10). This position approximates the ischemic boundary zone. The immunosuppressive compound, cyclosporin A (Novartis), was applied intraperitoneally immediately after this implantation and every other day before animals were killed with a 10 mg/kg dose.
Histochemistry
The surviving mice (Sham + PBS group, n = 3, MCAO + PBS group, n = 8; Sham + iPS group, n = 8; MCAO + iPS group, n = 7; Sham + virus-free iPS group, n = 3; MACO + virus-free iPS group, n = 4) were anesthetized by IP injection of ketamine hydrochloride, and then perfused with chilled phosphate-buffered saline (PBS), followed by 4% paraformaldehyde in 0.1 mol/L phosphate buffer. After postfixation overnight, 50-μm-thick sections were cut with a vibrating blade microtome (VT1000S; Leica). For immunohistochemistry, the following primary antibodies were used: rabbit anti-c-Myc antibody (1:100, Abcam); rabbit anti-Sox2 antibody (1:100, Santa Cruz Biotechnology); rabbit anti-Oct3/4 antibody (1:100, Santa Cruz Biotechnology); rabbit anti-Klf4 antibody (1:100, Lifespan Biosciences); rabbit anti-MMP-9 antibody (1:200; Chemicon), or rabbit anti-phospho-VEGF Receptor2 (Y1054) antibody (1:200, Abcam). The antibodies against c-Myc, Sox2, Oct3/4, and Klf4 were detected with secondary antibodies conjugated with Alexa Fluor™ (Molecular Probes). To estimate the expression of c-Myc, Sox2, Oct3/4, MMP-9, and phospho-VEGFR2, the sections were incubated with the respective first antibody and then with an appropriate biotin-labeled secondary antibody (1:500). After incubation with ABC Elite complex (Vector Laboratories), the signal was visualized with diaminobenzidine tetrahydrochloride.
Quantitative Analysis
For the semiquantitative evaluation of histochemical stainings, such as for c-Myc, Sox2, Oct3/4, MMP-9, and phospho-VEGFR2, the stained sections were selected from three levels of the caudate putamen (1.0, 0.5, and 0 mm rostral to bregma) (10) of each animal. Three areas in the peri-infarct cortex (Sham + PBS group and MCAO + PBS group) or in the iPS-derived tumors (Sham + iPS group, MACO + iPS group, Sham + virus-free iPS group, and MACO + virus-free iPS group) were chosen randomly in each section, and captured at 200× magnification with a microscope (BX51; Olympus). We confirmed the location of tumors or the border between the ischemic core and peri-infarct lesion through cresyl violet staining of adjacent sections. The staining intensity was measured using Scion Image software (Scion Corporation).
Statistical Analysis
Values are expressed as means ± SD. The frequencies of tumorigenesis were compared by an m × n chi-square test. The differences in tumor volume, intensity of c-Myc, Sox2, Oct3/4, MMP-9, and phospho-VEGFR2 staining, were evaluated for statistical significance by nonrepeated measures ANOVA and SNK test. In all statistical analyses, significance was accepted at p < 0.05.
Results
We prepared the murine iPS cells, which were established from Nanog-GFP transgenic mice (23), for transplantation. After having been cultured on mouse embryonic fibroblast for 10 days as described previously by Takahashi et al. (29), we confirmed that the majority of murine iPS cell colonies expressed the iPS cell marker, Nanog-GFP (Fig. 1A). The majority of these colonies also expressed stem cell markers, SSEA-1, nestin (data not shown), and four induced transcription factors, c-Myc, Sox2, Oct3/4, and Klf4 (Fig. 1B–E).

Murine iPS cells could express Nanog-GFP in the ischemic brain. (A–E) Murine iPS cells, iPS-MEF-Ng-20D-17, were made from Nanog-GFP transgenic mouse, and expressed Nanog-GFP, c-Myc, Sox-2, Oct3/4, and Klf4 in vitro culture. Scale bar: 50 μm. (F–I) iPS cells implanted into the ischemic striatum. Qtracker was added to iPS cells as a cell tracer, and the signal was detected 3 days after implantation (F). As time advanced, Nanog-GFP-positive iPS cells increased and extended into the ischemic striatum/cortex. At the same time, added Qtracker was diluted to be undetectable at 14 days after transplantation (G–I). The outlined areas in (I) indicate the fields shown in (H). Scale bars: 100 μm (F–H), 2 mm (I). n = 3 in each group.
In this study, we used a mouse model of middle cerebral artery occlusion (MCAO) (32), in which the infarcted region is mainly located in the lateral portion of the corpus striatum and cortex. To study whether the transplanted cells could survive in the ischemic striatum/cortex, we labeled iPS cells with a cell tracer, Qtracker, and transplanted these cells into the ipsilateral striatum/cortex 24 h after MCAO, and observed transplanted cells 3, 7, and 14 days after transplantation (n = 3 in each group). Three days after transplantation, we could detect Qtacker-positive transplanted cells in the striatum/cortex. At this time point, expression of Nanog-GFP was very low (Fig. 1F), although we observed that Nanog-GFP-positive transplanted cells increased and expanded into the ischemic striatum/cortex at 7 or 14 days after transplantation (Fig. 1G–I). Based on these finding, we designed the following experiments in which we studied the effect of ischemic condition against the fate of iPS cells.
We studied brain sections stained with cresyl violet, obtained from four experimental groups (Sham + PBS group, MCAO + PBS group, Sham + iPS group, MACO + iPS group) to evaluate the incidence of tumorigenesis, and tumor volume. iPS cells formed tumors in all cases of the MCAO + iPS group (day 14, n = 5; day 28, n = 6) and tumors were significantly larger than those of the Sham + iPS group (*p < 0.05) (Table 1). We transplanted iPS cells in the striatum/cortex, but the iPS cells formed tumors only around the needle tracks of the cortex in the Sham + iPS group (Fig. 2B). In contrast, iPS cells spread out over the ischemic area and formed large tumors occupying infracted regions in the MCAO + iPS group (Fig. 2B). However, these cells scarcely penetrated the nonischemic, intact brain area, similar to the Sham + iPS group, and stayed within the ischemic area (Fig. 2C, D). Hematoxylin-eosin staining revealed that transplanted iPS cells formed a teratoma both in the Sham + iPS group and in the MCAO + iPS group (Fig. 3).

iPS cells spread, not to the intact area, but to the infarcted lesions. (A–D) Coronal brain sections obtained 14 or 28 days after transplantation, and stained with cresyl violet. (A) Focal cerebral ischemia without iPS cell transplantation obviously reduced the number of cells stained with cresyl violet in the ipsilateral side. Dotted lines indicate the border between the intact and infracted areas. Scale bar: 2 mm. (B) Transplanted iPS cells did not penetrate into the intact area, and stayed around the injection site in the sham-operated brain. (C, D) iPS cells spread over the infracted area and formed tumors squeezing the intact area in the postischemic brain.
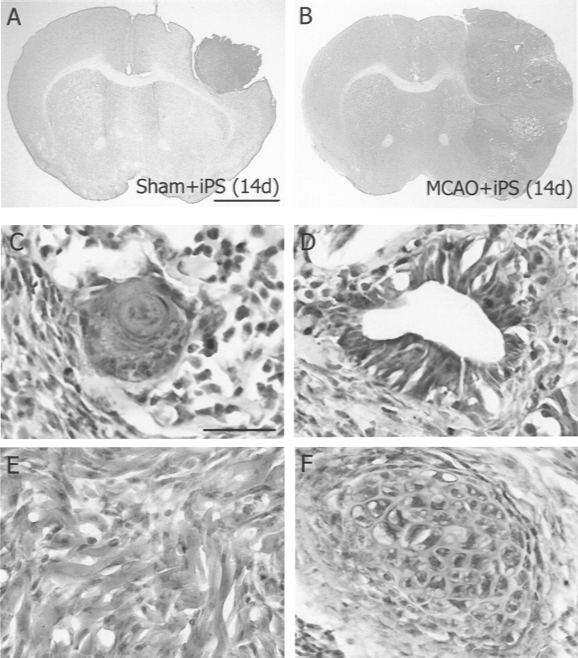
Undifferentiated iPS cells formed teratoma. Coronal brain sections obtained from Sham + iPS group (A) and MCAO + iPS group (B–F) 14 days after transplantation, and stained with hematoxylin-eosin. The iPS-derived tumors included ectodermal cells with cornification (C), cylinderlike epithelium cells (D), striated muscle-like cells (E), and chondrocyte (F). Scale bars: 2 mm (A), 50 μm (C).
Incidence of Tumorigenesis and Tumor Volume After Intracerebral Implantation of Murine iPS Cells

Values are means ± SD. iPS cells implanted into the ischemic brain formed tumors with higher probability and larger volume than those implanted into the sham-operated brain.
p < 0.05 (m × n, chi-square test).
p < 0.01 (nonrepeated measures ANOVA and SNK test).
Our results indicate that iPS cells can increase more rapidly in the postischemic condition. The iPS cell line was produced from mouse fibroblasts by retroviral introduction of c-Myc, Sox2, Oct3/4, and Klf4 (23). It has been reported that reactivation of the c-Myc increases tumorigenicity in germline-competent chimeras produced by murine iPS cells (20). We then evaluated whether the expression of these four transcription factors changed in the postischemic brain. To our surprise, not only c-Myc but also Sox2 and Oct3/4 expression was significantly increased in the tumors of the MCAO + iPS group, while the expression of Klf4 was low (*p < 0.05, **p < 0.01) (Fig. 4).

Cerebral ischemia might activate c-Myc, Sox2, and Oct3/4 in tumors. (A–D) Immunohistochemical staining for c-Myc, Sox2, Oct3/4, and Klf4 in the iPS-derived tumors. The insets in (A–D) indicate the fields of each magnified images, respectively. Scale bar: 2 mm (20 μm, magnified image). (E–G) Relative intensity of c-Myc, Sox2, and Oct3/4 in the peri-infarct lesion of cortex or iPS-derived tumors. The expression of c-Myc, Sox2, and Oct3/4 were evidently increased in iPS-derived tumors (*p < 0.05, **p < 0.01).
Recent studies reported that these four transcriptional factors, which are integrated into the genome by retrovirus vectors, can continue to express in iPS cells, alter their characteristics, and also induce tumorigenesis (27). We then transplanted another iPS cell line, which was established without a retrovirus vector and did not have an integrated exogene (24), into the ischemic or sham-operated brain, and evaluated the incidence of tumorigenesis, and tumor volume at 28 days after transplantation. We found tumors in all transplanted brains (Sham + virus-free iPS group, n = 3; MCAO + virus-free iPS group, n = 4), but there was no significant difference in tumor volume between the two groups (Fig. 5). These results suggest that integrated transcriptional factors might accelerate teratoma formation in the ischemic brain.

A virus-free mouse iPS cell line formed tumors, but tumor volume were not increased by cerebral ischemia. Murine iPS cells, iPS-MEF-Ng-492B-4, which were established without retrovirus vector, were also transplanted into the sham-operated mouse brain (n = 3) or the ischemic brain (n = 4). (A, B) Coronal brain sections obtained 28 days after transplantation and stained with cresyl violet. (C) There was no significant difference in the tumors volumes between the Sham + virus-free iPS group and the MCAO + virus-free iPS group.
Recent cancer research has shown that c-Myc can activate VEGFR2 expression, promoting tumor-related angiogenesis. In conjunction with hypoxia, c-Myc activation further promotes the VEGF signal (12). c-Myc activation has also been reported to induce MMP-9 expression (15). MMP-9 was expressed in the ischemic murine brain (26) or in human neuroblastoma (3), and MMP-9 can play an important role in neural precursor migration (14) by degrading the extracellular matrix (19). The above results and reports led us to hypothesize that the integrated transcriptional factors and ischemic condition might induce MMP-9, or activate the VEGF signal in the MCAO + iPS group. We therefore examined histological sections stained with an anti-MMP-9 antibody or an anti-phospho-VEGFR2 antibody. Both MMP-9 and phospho-VEGFR2 were abundantly expressed not only in infracted regions but also in iPS-derived tumors 14 days after transplantation (Fig. 6A–G, I–O). Semiquantitative analysis of MMP-9 and phospho-VEGFR2 staining showed that MMP-9 and phospho-VEGFR2 expression increased significantly in the MCAO + iPS group compared with the Sham + PBS group or the Sham p+ iPS group (*p < 0.05, **p < 0.01) (Fig. 6H, P). These results indicate that MMP-9 and VEGF signals, which can be induced by activated c-Myc and cerebral ischemia, might play an important role in the expansion of iPS-derived tumors.

Both MMP-9 and phospho-VEGFR2 were abundantly expressed in iPS-derived tumors. Immunohistochemical staining for MMP-9 (A–G) and phospho-VEGFR2 (I–O) in the peri-infarct lesion of cortex or iPS-derived tumors 14 days after transplantation. Co, cerebral cortex; Cc, corpus callosum. The insets in (B, C, D, J, K, L) indicate the fields shown in (E, F, G, M, N, O) respectively. Scale bars: 250 μm (A–D, I–L). Scale bars: 20 μm (E–G, M–O). Some astrocyte-like cells expressed MMP-9 (arrows, F), while iPS-derived tumor cells itself expressed MMP-9 (E, G). Relative intensity of MMP-9 (H) and phospho-VEGFR2 (P) in the peri-infarct lesion of cortex or iPS-derived tumors at 14 or 28 days. The expression of MMP-9 and phospho-VEGFR2 were significantly increased in the MCAO + iPS group compared with Sham + PBS group or Sham + iPS group (*p < 0.05).
Discussion
In this article, we showed that an iPS cell line established with a retrovirus multiplied and expanded rapidly within the postischemic region of the mouse brain. On the other hand, a virus-free iPS cell line formed tumors, but the ischemic condition did not increase tumor size (Fig. 5). We also showed that the tumors, which were derived from the iPS cells established with retrovirus, highly expressed c-Myc, Sox2, and Oct3/4 in ischemic brain (Fig. 4), suggesting that integrated transcriptional factor might affect cell characteristics and enhance the outgrowth of transplanted iPS cells under the ischemic condition. Among four transcriptional factors, c-Myc in particular has been studied as an oncogene. c-Myc can boost global gene expression and influence a wide spectrum of cellular pathways by acting on a large number of genes, including MMP-9 and VEGFR2 (6). We found that both MMP-9 and phospho-VEGFR2 were strongly expressed in iPS cells transplanted into the ischemic brain (Fig. 6). MMP-9 can promote tissue remodeling by degrading the extracellular matrix. Activated VEGFR2 can increase angiogenesis. We cannot exclude the possibility that other genes also affected tumor size, but our findings suggested that both MMP-9 and activated VEGF signals could take part in forming larger tumors. In addition, NGF and EGF have been reported to activate c-Myc (8). LIF signals preferentially activated Sox2 and Oct3/4 through the Jak-Stat pathway, to maintain pluripotency in mouse ES cells (22). Various cytokines such as NGF (9), EGF (21), FGF2, LIF (28), and VEGF (16) are known to be induced in the ischemic brain. Therefore, these cytokines might activate transcriptional factors, maintain pluriopotency of iPS cells, increase cell number, and accelerate the growth of tumors in the ischemic brain.
Our data showed a possibility that pathologic in vivo environment may raise tumorigenicity or increase tumor size of transplanted iPS cells. Cell transplantation into postischemic brain can be useful and sensitive experimental tools to check whether the iPS cells can make tumors in vivo. We have to check the safety of iPS cells not only in the normal, healthy animal model, but also in the pathologic, disease animal model in order to develop a novel therapeutic strategy for various neurological disorders.
Footnotes
Acknowledgments
We are grateful to RIKEN Biore-source Center for Murine iPS cell lines. This work was partly supported by Grant-in-Aid for Scientific Research (B) 21390267 and the Ministry of Education, Science, Culture and Sports of Japan, and by Grants-in-Aid from the Research Committee of CNS Degenerative Diseases (Nakano I), and grants (Itoyama Y, Imai T) from the Ministry of Health, Labour and Welfare of Japan.