
Abstract
Adult olfactory ectomesenchymal stem cells (OE-MSCs) and olfactory ensheathing cells (OECs), both from the nasal olfactory lamina propria, display robust regenerative properties when transplanted into the nervous system, but the mechanisms supporting such therapeutic effects remain unknown. Matrix metalloproteinases (MMPs) are an important family of proteinases contributing to cell motility and axonal outgrowth across the extracellular matrix (ECM) in physiological and pathological conditions. In this study, we have characterized for the first time in nasal human OE-MSCs the expression profile of some MMPs currently associated with cell migration and invasiveness. We demonstrate different patterns of expression for MMP-1, MMP-2, MMP-9, and MT1-MMP upon cell migration when compared with nonmigrating cells. Our results establish a correspondence between the localization of these proteinases in the migration front with the ability of cells to migrate. Using various modulators of MMP activity, we also show that at least MMP-2, MMP-9, and MT1-MMP contribute to OE-MSC migration in an in vitro 3D test. Furthermore, we demonstrate under the same conditions of culture used for in vivo transplantation that OE-MSCs and OECs secrete neurotrophic factors that promote neurite outgrowth of cortical and dorsal root ganglia (DRG) neurons, as well as axo-dendritic differentiation of cortical neurons. These effects were abolished by the depletion of MMP-2 and MMP-9 from the culture conditioned media. Altogether, our results provide the first evidence that MMPs may contribute to the therapeutic features of OE-MSCs and OECs through the control of their motility and/or their neurotrophic properties. Our data provide new insight into the mechanisms of neuroregeneration and will contribute to optimization of cell therapy strategies.
Keywords
Introduction
The olfactory nasal tissue is a paradigm of continuous remodeling in the nervous system. The mucosa includes two tissues separated by a basal lamina: (i) the epithelium, which harbors the olfactory sensory neurons, and (ii) the lamina propria, which is home for the ensheathing cells that nourish and guide the growing axons during their migration towards the olfactory bulb. Both compartments contain a population of stem cells with specific characteristics: bipotent epithelial stem cells in the epithelium and multipotent mesenchymal-like stem cells in the lamina propria (28). We have recently characterized the human lamina propria stem cells as a subtype of mesenchymal stem cells with neurogenic and osteogenic properties and named them olfactory ectomesenchymal stem cells (OE-MSCs) (6). OE-MSCs are a potential source of cells for autologous cell transplantation therapy in Parkinson's disease (30), hearing loss (34), and lesion-induced amnesia (31).
OECs are also known for their therapeutic properties. They have been successfully assessed in animal models of spinal cord injury, stroke, degenerative diseases, and peripheral nerve trauma [for review, see (43)]. Following convincing preclinical tests in rodents (26, 27), clinical studies based on autologous transplants of OECs in paraplegic patients were performed (9, 25). Although these findings highlight the clinical potential of olfactory nasal olfactory cells, the underlying molecular mechanisms, notably those supporting migration of OE-MSCs and the neurotrophic effects of both OE-MSCs and OECs remain largely unknown.
Matrix metalloproteinases (MMPs) form a multigenic family of Zn2+-dependent endopeptidases that belong to the superfamily of metzincins. MMPs can be roughly classified into secreted and membrane-type MMPs (MT-MMPs) (29). The four secreted tissue inhibitors of MMPs (TIMP-1 to TIMP-4) control MMP activity via the interaction of their N-terminal domain with the catalytic domain of the enzymes. In addition to the inhibition of MMPs, the TIMPs display pleiotropic effects in a wide range of cell types (42). MMPs regulate the pericellular environment in physiological and pathological conditions through the proteolytic processing of extracellular matrix (ECM) and signaling proteins (e.g., cytokines, chemokines, cell adhesion molecules, membrane receptors, etc.) (8). For instance, MMPs may mobilize growth and angiogenic factors entrapped in the ECM via proteolytic-mediated release (4) and also catalyse the conversion of inactive forms of these factors into their biologically active forms (23). For these reasons, MMPs are pivotal for fundamental cell functions, including cell migration in physiological and pathological conditions. Some MMPs such as MMP-1, -2, -9 or MT1-MMP have been extensively studied as promoters of tumor cell invasion and metastasis, but also as mediators of postlesion tissue repair [for review (7, 14, 22, 37, 38)]. In the nervous system, increasing experimental evidence identifies MMPs as regulators of neural cell motility in physiological or pathological tissue remodeling (37). For instance, the early postnatal migration of cerebellar granule cell precursors in mice parallels changes in MMP-9 expression, and the genetic- or antibody-mediated inhibition of the enzyme results in a delay in cerebellar external granular layer migration (44). Likewise, the inhibition of furin, the enzyme that catalyzes the activation of MT-MMPs, inhibits the physiological migration of neuroblasts along the rostral migratory stream (RMS), suggesting the implication of MT-MMPs in this process (3). In the postischemic brain, neuroblasts that migrate from the subventricular zone (SVZ) into the injured striatum exhibit upregulated levels of MMP-9 and broad spectrum inhibitors of MMPs interfere with this migration (24). MMPs also influence the motility of differentiated neural cells; we and others have shown that MMP-2 promotes migration of cultured resting astrocytes, possibly via interaction with β1 integrin and the actin cytoskeleton (32), whereas MMP-9 contributes to astrocyte motility during glial scar formation (20). More recently, we have also shown that MMP-2 and MMP-9 support motility of cultured OECs from the nasal mucosa (18). From the above, it follows that cells, including grafted cells, could use MMPs to surmount natural barriers and boost neural plasticity and regeneration.
Accordingly, we have characterized at the subcellular level the expression profile and distribution of MMPs potentially involved in OE-MSCs migration, and we investigated their role in cell motility. Moreover, based on recent data implicating MMPs in neurite outgrowth and axo-dendritic differentiation of cortical neurons (16, 33) and neurogenesis (46), we also investigated the possible trophic effects of MMPs secreted by OE-MSCs and OECs on cortical and dorsal root ganglia (DRG) neurons. Our data demonstrate the role of MMPs in all these phenomena and suggest their contribution to the neuro-regenerative properties of grafted nasal olfactory cells.
Materials and Methods
All the procedures used to generate cell cultures in the present work from human beings or animals were approved by the Ethics Committee of the Medical Faculty of Marseilles and conform to National and European regulations (EU Directive Nu 86/609).
Culture of Human OE-MSCs and BMSCs
Human OE-MSCs and human bone marrow mesenchymal stem cells (BMSCs) were collected for a previous study from two males and two females with age ranging from 23 to 52 years old with ethical approval and informed consent (6), and aliquots of early passaged cells from all four patients were used for the current study. In proliferative conditions, stem cells were cultivated in Dulbecco's modified Eagle's medium (DMEM)/HAM'S F12 supplemented with 10% fetal bovine serum (FBS), 1% penicillin, and 1% streptomycin (all from Gibco, Invitrogen, Carlsbad, CA, USA).
Culture of Rat Olfactory Ensheathing Cells
All experiments were conducted on adult (6–8 weeks old), pathogen-free Lewis female rats (Elevage Janvier, Le Genest Saint Isle, France). Rats were deeply anesthetized by intraperitoneal injection of 1 ml of Lethabarb (sodium pentobarbital 54.7%, Ceva Santé Animale, Libourne, France). All efforts were made to minimize animal suffering and reduce the number of animals used. After death, rats were decapitated. The olfactory mucosa was exposed and excised according to the recently described procedure (15). The tissue was immediately placed in 37°C DMEM/F12 with GlutaMAX” (Gibco), washed two times, and incubated for 45 min at 37°C in 1 ml of an enzymatic solution of Dispase II (2.4 units/ml, in Puck's solution; Roche Diagnostics, Penzbeg, Germany). A microspatula (Harvard Apparatus, Holliston, MA, USA) was used to gently separate the olfactory epithelium from the lamina propria under a dissection microscope. The lamina propria was then placed for 10 min at room temperature in 1 ml of Hank's balanced salt solution (HBSS, calcium and magnesium free, Invitrogen) to inactivate Dispase II, transferred to 1 ml of collagenase I (0.25% in serum-free DMEM/F12; Sigma-Aldrich, St. Louis, MO, USA), and incubated for 5–10 min at 37°C/5% CO2. The lamina propria was mechanically triturated with the help of a Pasteur pipette (Dominique Dutscher, Brumath, France), at the start and at the end of the incubation period. The cell suspension was transferred to a 15-ml conical tube (Falcon, BD Biosciences, Franklin Lakes, NJ, USA), and collagenase activity was inhibited by adding 9 ml of HBSS. After centrifugation at 400 × g for 5 min, the supernatant was aspirated and the cell pellet was resuspended in DMEM/F12 supplemented with 10% FBS. A viable cell count was made with a Malassez cell (Dominique Dutscher), and cells from the lamina propria were plated in 25 cm2 flasks and maintained for 15 days at 37°C and 5% CO2. The growth medium was replaced with fresh medium every 2 days. Cells were then split by trypsinization and replated at 1 × 105 cells/cm2 in an equal final volume of 500 μl of serum-free DMEM/F12 supplemented with 25 ng/ml of transforming growth factor-α (TGF-α, Sigma-Aldrich) onto glass coverslips precoated with 2 μg/cm2 of poly-l-lysine (Sigma-Aldrich) in culture dishes (Millipore, Bellirica MA, USA). The second passage was performed 1 week after.
Primary Cultures of Neurons
Primary cultures of cortical neurons were prepared from embryos of CD1 mice bred in our facilities, as previously reported (33). Pregnant females were deeply anesthetized with halothane (Nicholas Piramal Limited, London, UK). E17–18 embryos were quickly removed and put into cold HBSS containing 0.5% glucose (both from Invitrogen). Following decapitation, the cerebral cortices were dissected, pooled, and enzymatically dissociated for 10 min at 37°C in HBSS containing 0.1% trypsin (Invitrogen) and 10 μg/ml DNase I (Sigma-Aldrich). Adding HBSS containing 5% FBS stopped the enzymatic reaction. Further mechanical dissociation was carried out in HBSS containing 0.05% DNase by trituration through a Pasteur pipette. After 5 min centrifugation at 300 × g, the cell pellets were resuspended in the plating medium containing MEM, 0.6% glucose, 1 mM sodium pyruvate, 5 U/ml penicillin/streptomycin, and 10% FBS (all from Invitrogen). Cells were plated in 13-mm diameter wells (Millipore), onto 12-mm glass coverslips precoated with 1 mg/ml poly-d-lysine (Sigma-Aldrich) in borate buffer pH 8.5, and were grown at 37°C in a humidified atmosphere containing 5% CO2. After 75 min, the plating medium was aspirated and replaced by serum-free defined medium consisting of Neurobasal, with B27 supplement, 5 U/ml penicillin/streptomycin, 2.5 mM l-glutamine, and 25 nM glutamate (all from Invitrogen).
Cultures of DRGs
CD1 mouse pups (postnatal day 2 or 3) were deeply anesthetized with halothane; the DRGs were dissected in DMEM and plated during 12 h in four-well plates on 12 mm poly-l-lysine/laminin-coated cover slips (Sigma-Aldrich) in DMEM. After 12 h, conditioned DMEM was removed and replaced by fresh or 72 h OEC-conditioned neurobasal media, containing B27 supplement, 5 U/ml penicillin/streptomycin, 2.5 mM l-glutamine, and 25 nM glutamate (all from Invitrogen), with or without gelatinases.
Modulation of MMP Activity
The following MMP inhibitors were added to serum-free culture media: (1) human recombinant truncated N-terminal form of TIMP-1 (N-TIMP-1; Hideaki Nagase) (19) with similar Ki values (0.2–0.4 nM) for MMP-1, MMP-2, MMP-3, and MMP-9 and 146 nM for MT1-MMP; (2) MMP selective pseudophosphinic RXPO3R inhibitor (Dr. Dive) that does not inhibit adamalysins (5, 33), with Ki for MMP-2, MMP-9, and MT1-MMP of 55, 41, and 91 nM, respectively; (3) MMP and adamalysin inhibitor GM6001 (Sigma-Aldrich) with Ki = 500 pM for MMP-2, Ki = 27 nM for MMP-3, Ki = 100 pM for MMP-8, and Ki = 200 pM for MMP-9; (4) selective MMP-2 inhibitor (MMP-2 inhibitor III, Calbiochem, La Jolla, CA, USA), which exhibits good selectivity for MMP-2 (Ki 12 nM) when compared with MMP-9 and MMP-3 (Ki 200 and 4500 nM, respectively); (5) MMP-9 inhibitor-I (Calbiochem) (IC50 = 5 nM); and (6) MT1-MMP neutralizing antibody (LEM-2/15; Alicia Garcia Arroyo) (13). MMP-2 and MMP-9 selective inhibitors were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) at a final concentration of 0.04%. Controls were incubated with the same concentration of DMSO for synthetic inhibitors or IgG for anti-MT1-MMP. A range of concentrations based on values of IC50 provided by the manufacturer was used to determine the concentrations of selective MMP-2 and MMP-9 inhibitors used in Boyden chamber-based cell migration assay (Millipore). The lowest efficient concentration was used.
The Silicone Stencil Assay for Synchronized Cell Polarization/Migration
The silicone stencil system made from poly (dimethylsiloxane) (PDMS; Sigma) was set up in order to study the behavior of migrating cells. We adapted previous methods (10, 36) to our specific experimental conditions. We manufactured reusable circular silicone inserts of defined diameter containing an internal rectangular stencil of 15 mm2 where the cells were plated, on four-well tissue culture plates (Nunc, Roskilde, Denmark). Manufacture of the inserts consisted in mixing in a plastic pillbox (VWR, West Chester, PA, USA) 12 g base with 3 g of curing agent (Down Corning Corporation, Midland, MI, USA) to obtain a silicone elastomer containing 20% of curing agent. The mixture was degassed for 15–20 min using a vacuum speed device. After degassing, the mixture was poured into a Petri dish (BD Biosciences) and first heated for 60 min at 80°C followed by an additional crosslinking step at 80°C for 60 min. After 1 h at room temperature to obtain the polymer material, silicon chips were cut using a copper tube and customized rectangular miniblades. Sterilized supports were inserted into the wells, and the plates were incubated for 30 min at 37°C to allow the silicone inserts to attach to the plate surface. The stencil was loaded with 200 μl of culture medium containing 4 × 103 OE-MSCs (666 mm2). After 24 h, adherent cells formed a confluent monolayer and the stencil was removed using forceps. Nonadherent cells were removed following two washes with serum-free medium. Subsequently, 500 μl of fresh culture DMEM/HAM'S F12 supplemented with 1% ITS (insulin, transferrin, selenium; Invitrogen), 1% penicillin, and 1% streptomycin were added to the cells.
Boyden Chamber-Based Cell Migration Assay
Confluent OE-MSCs cultured in 25 cm2 flasks were detached by trypsin/EDTA, counted, and seeded into the upper chamber of transwell polyethylene terephthalate filter membranes with 8-μm diameter pores (Corning, Chorges, France) at a density of 2 × 104 cells/well, in a final volume of 300 μl serum-free medium containing 1% ITS. Cells were allowed to migrate through the membrane filter for 24 h in the absence or presence of RXPO3R, N-TIMP-1, MMP-2I, MMP-9I, and LEM-2/15 in the upper and lower chambers. Cells migrating through the membrane pore and invading the underside surface of the membrane were fixed with 4% paraformaldehyde (PFA) (Sigma-Aldrich). Nonmigratory cells on the upper membrane surface were removed with a cotton swab, and nuclei were stained with 0.5 μg/ml DNA intercalant Hoechst #33 258 (Invitrogen). Filters were observed using a Nikon E800 upright microscope equipped with epifluorescence, tetrarhodamine isothiocyanate (TRITC), fluorescein isothiocyanate (FITC), and 4′,6-diamidino-2-phenylindole (DAPI) filters (Nikon, Champigny-sur-Marne, France) and an Orca-ER CCD camera (Hamamatsu Photonics, Hamamatsu, Japon). For quantitative assessment, the number of stained migrating cells was counted with ImageJ software (NIH, Bethesda, MD, USA) on 12 random fields per membrane filter at 10 × magnification.
Isolation of Intracellular, Cytoskeletal, and Membrane Fractions
Subcellular fractions were obtained from cultured OE-MSCs using the ProteoExtract subcellular proteome extraction kit (Calbiochem) according to the manufacturer's instructions. Proteins were extracted with different buffers from major subcellular compartments and on the basis of the difference in protein solubility. The first cytosolic fraction was obtained by incubating the cells with an extraction manufacturer's buffer at 4°C for 10 min. A second fraction containing cell membranes was obtained by incubating the cells with an extraction buffer for 30 min at 4°C. The nucleus and cytoskeleton fractions were also obtained by the same procedure. The enrichment of each fraction was evaluated by probing the Western blots with specific antibodies against CD44 (Millipore, Billerica, MA, USA) for the membrane fraction, β-actin (Sigma-Aldrich) for the cytoskeleton fraction, and histone-H3 (Sigma-Aldrich) for the nuclear fraction. The different subcellular fractions were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to Western blot or gel zymography (see below).
Gelatin and Collagen Gel Zymography
Gel zymography was performed as previously described (33). Briefly, culture supernatants or subcellular fractions were collected, and protein concentrations were normalized using the Lowry method (Bio-Rad, Hercules CA, USA). Equal amounts of protein were subjected to 8.5% SDS-PAGE (Bio-Rad) containing 4.5 mg/ml of porcine gelatin (Sigma-Aldrich) or 1 mg/ml of native collagen type I (Sigma-Aldrich) in nondenaturing and non-reducing conditions. Gels were washed twice for 30 min in 2.5% Triton X-100 (Sigma) to remove SDS and incubated for 48 h in 50 mM Tris (pH 7.5), 10 mM CaCl2 (Sigma-Aldrich) at 37°C. Gels were then stained with 0.1% Coomassie blue G-250 (Sigma-Aldrich) for 3 h in 30% ethanol and distained with a solution containing 5% acetic acid (Sigma-Aldrich) until clear bands of gelatinolysis appeared on a dark background. Gels were digitized, and optical densities were assessed with the Bio 1D software (Vilber Lourmat, Marne-la-Vallée, France). Human recombinant active MMP-2 (300 pg) (Millipore, Malscheim, France) was used as a positive and normalizing control.
Extraction of Gelatinases
All procedures were performed at 4°C. Culture supernatants from OE-MSCs or OECs were collected after 72 h in neurobasal medium. For gelatinase extraction, 5 ml of supernatant were incubated 24 h with 5 ml of gelatin-sepharose 4B (Amersham Bioscience, Buckinghamshire, UK) with constant shaking. After centrifugation, the supernatant was placed at −80°C, and the gelatin sepharose pellet was resuspended in working buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 5 mM CaCl2, 0.005% Brij-35; all from Sigma-Aldrich) and the solution was centrifuged again. The pellet was then incubated for 30 min with elution buffer consisting of working buffer containing 10% DMSO. The efficacy of gelatinase extraction was measured by gel zymography.
Western Blot
Primary Antibodies and Other Probes Used in Immunocytochemistry and Western Blot Assays

MMP, matrix metalloproteinases; MT1, membrane type 1; MAP-2, microtubule-associated protein-2.
Immunocytochemistry
OE-MSCs, cortical neurons, or DRGs were rinsed three times in phosphate-buffered salt (PBS) and fixed in 4% PFA for 20 min. For immunocytochemistry, cells were preincu-bated with 0.1% Triton X-100, 3% bovine serum albumin (BSA) (all from Sigma-Aldrich) for 60 min, followed by 90 min incubation at room temperature with mouse monoclonal anti-βIII tubulin, (Sigma-Aldrich), mouse monoclonal anti-Tau-1 (Sigma-Aldrich), rabbit anti-microtubule associated protein-2 (MAP-2; Abcam, Cambridge, UK), rabbit anti-MMP-9 (Millipore), mouse anti-MMP-1 (EMD Chemicals), rabbit anti-MMP-2 (Millipore), and rabbit anti-MT1-MMP (Epitomics). The secondary antibodies were labeled with Alexa Fluor® (Invitrogen), and all secondary antibodies were at a concentration of 1/800 (for further details see Table 1). The antibodies were diluted in PBS containing 0.1% Triton X-100, 3% BSA. Cells were rinsed three times in PBS and incubated for 1 h with Alexa Fluor® 488-594 antibodies (Invitrogen) and 0.5 μg/ml of nuclear marker Hoechst #33258. For cytoskeleton labeling (F-actin), cells were incubated after immunocytochemistry with Texas Red-X phalloidin (Sigma-Aldrich) for 1 h at room temperature, then rinsed in PBS, and mounted in fluorescence mounting medium (Dako, Glostrup, Denmark). Cells were observed under a LSM 700 confocal microscope (Zeiss Jena, Germany). Images were analyzed using the ZEN (Zeiss) and ImageJ software.
In Situ Zymography on FITC-Labeled Gelatin
In order to localize net gelatinolytic activity on OE-MSCs, we used an in situ zymography method previously described (32, 39). Briefly, OE-MSCs were grown on glass coverslips, and the medium was supplemented to a final concentration of 5 mM CaCl2 and 10 μg/ml of FITC-labeled DQ™ gelatin intramolecularly quenched (EnzCheck Collagenase kit from Invitrogen). After 1 h at 37°C in a humidified atmosphere containing 5% CO2, cells were rinsed in PBS, fixed with 4% PFA for 5 min, and processed for immunocytochemistry for MMPs and nuclear labeling, as stated above. Cell images were acquired with a LSM 700 confocal microscope and analyzed with the ZEN software.
RNA Extraction, RT-PCR, and TaqMan Quantitative RT-PCR Analysis
Total RNA was prepared from cultures of OE-MSCs with the RNeasy Lipid Tissue Mini kit (Qiagen, Courta-boeuf, France). Single-strand cDNA for a template was synthesized from 1 mg of total RNA using primer oligo (dT)12–18 (Invitrogen) and moloney murine leukaemia virus (MMLV) reverse transcriptase (Invitrogen) using manufacturer recommendations.
qPCR experiments were carried out with the 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). All reactions were performed using TaqMan Fast Universal PCR Master Mix and the following probes TaqMan® Gene Expression Assays according to the manufacturer's instructions (Applied Biosystems): MMP-1 Hs00899658_m1, MMP-2 Hs01548727_m1, MMP-9 Hs00234579_m1, MT1-MMP Hs00237119_m1, TIMP-1 Hs00171558_m1, and glyceraldehyde 3 phosphate dehydrogenase (GAPDH) Hs03929097_g1*. Twenty-five nanograms of previously prepared OE-MSC cDNA were used for each experiment. Samples were run in duplicates on the same 96-well plates and analyzed using the 7,500 Software v2.0 (Applied Biosystems). Thermal cycling conditions started with initial denaturation at 95°C for 20 s, followed by 40 cycles of denaturation at 95°C for 3 s, and annealing/extension at 60°C for 30 s. Relative expression levels were determined according to the ΔΔCT method. In the latter, gene expression level was quantified using the following formula: 2-ΔΔCT, where ΔΔCT = ΔCT target gene – ΔCT reference gene (GAPDH) in the same sample (2). All values were normalized with respect to the 6 h time point.
Cell Viability Assay
Cell viability was assessed using the 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Sigma-Aldrich), which measures mitochondrial activity in living cells. Data were calculated as the percentage of living cells = 100 – [(treated cells OD550/tcontrol cells OD550) × 100].
Morphological Analyses of Cortical and DRG Neurons
In each experiment with cortical neurons, fluorescence microphotographs of at least five randomly selected fields per well and three wells per experimental condition were taken. The following parameters were analyzed: (i) total number of cells, determined on the basis of nuclear Hoechst staining, and the MAP-2/Tau-1 ratio from doublelabeled samples and (ii) neurite length of all MAP-2 and Tau-1-positive cells was manually drawn using a computer mouse. The mean length of the neuritic arbor was obtained by dividing the total length of neurites in a field by the number of cells immunoreactive for MAP-2 and Tau-1. A neurite segment was defined as the distance between branching points or the distance between a branching point and the tip of the neurite. The axon is defined as a neurite with a robust immunostaining for Tau-1, whereas MAP-2 labels all neurites. In order to evaluate the axo-dentritic differentiation, we determined the following ratio: number of neurites with a robust Tau-1 immunostaining/number of neurites with MAP-2 immunostaining.
DRG explants cultured for 3 days were fixed in 4% PFA and stained with the Tau-1 antibody. The area occupied by the axonal arbor was measured and normalized by the area of the explant using the LUCIA software (Laboratory Imaging, Prague, Czech Republic). Between five and seven explants per experimental group were analyzed in each independent experiment.
Statistical Analysis
Data were analyzed using analysis of the variance (ANOVA), followed by post hoc Tukey's test. The mean values ± SEM were obtained from at least three independent experiments, unless otherwise indicated. Statistical significance was achieved at p < 0.05.
Results
MMP-1, MMP-2, MMP-9, MT1-MMP, and TIMP-1 Are Expressed and Secreted by OE-MSCs (Fig. 1)
qRT-PCR was performed on cDNA prepared from total OE-MSC RNA, with MMP-1, MMP-2, MMP-9, MT1-MMP, and TIMP-1-specific probes (Fig. 1A). All mRNAs were detected in confluent OE-MSCs and remained relatively stable across time after explantation (Fig. 1A).
Study of MMP expression, secrection, and activity in OE-MSCs. (A) Histograms representing mRNA expression in fold time changes normalized with respect to the 6-h time point for matrix metalloproteinases (MMPs) and tissue inhibitor of MMP-1 (TIMP-1) following qRT-PCR on olfactory ectomesenchymal stem cells (OE-MSCs) after 6, 12, and 24 h in culture. The mRNA of the house-keeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as reference gene. The values represent the means ± SEM of three independent experiments. No significant differences were observed across experimental groups. (B) Collagenbased gel zymography, showing expression of various collagenolytic activities including both active and inactive forms of MMP-9 and MMP-2 and the active form of MMP-1 in OE-MSC supernatant. No signal for MMP-1 was detected in the supernatants from BMSCs. (C) Cell fractionation of OE-MSCs followed by gelatin-based gel zymography analysis showing the presence of MMP-2 at the size of the human recombinant active MMP-2 (lane 1) and pro- and active MMP-9 forms in all fractions. (D) Western blot analysis of supernatant and cell fractions from OE-MSCs showing MMP-2 as a doublet of 68–72 kDa corresponding to MMP-2 and pro-MMP-2 forms in the supernatant and all cell fractions, except for the cytoskeleton that exhibits the pro-MMP-2 only. MMP-9 appears as a doublet of 88–100 kDa corresponding to the MMP and pro-MMP-9 forms in the cytosolic fraction only. (E) Supernatant from OE-MSCs blotted with a specific anti-MMP-1 antibody showing two bands at 45–55 kDa probably corresponding to active and latent forms of MMP-1, respectively. (F) Cell lysate and cell fractionation followed by Western blot analysis with a specific anti-membrane type 1 (MT1)-MMP antibody showing the presence of MT1-MMP (60 kDa) in all fractions except the cytosol, and a relative enrichment of MT1-MMP in the membrane fraction. (G) Enrichment of cell fractions from OE-MSCs was assessed by Western blot on cytoskeletal, membrane and nuclear fractions using specific antibodies against actin, CD44, and histone H3, respectively.
We next used gel zymography to analyze MMP-1, MMP-2, and MMP-9 protein expression and activity in serum-free media conditioned by OE-MSCs for 48 h. A previous transcriptome analysis (6) revealed that MMP-1 mRNA was among the most differentially increased genes in OE-MSCs when compared with BMSCs (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24598). We sought further confirmation of these data using collagen gel zymography, and we detected a ~50-kDa band characteristic of MMP-1. A collagenolytic band of the expected molecular size was detected in medium conditioned by the OE-MSCs, whereas it was undetectable in medium conditioned by BMSCs (Fig. 1B). The protein was also detected by Western blot in the supernatant (Fig. 1E). Using the same zymographic technique, we detected in the supernatant several gelatinolytic bands corresponding to MMP-2 and MMP-9 (Fig. 1B). For MMP-2, we observed only a band representing the pro-form of MMP-2. MMP-9 is expressed as a doublet of 105 and 92 kDa that likely corresponds to the latent and active forms of the proteinase.
In order to determine the subcellular distribution of MMP-1, MMP-2, and MMP-9, we analyzed by gelatin zymography the major subcellular compartments (cytosolic, membrane/organelle, nuclear and cytoskeletal fractions) (Fig. 1C). We failed to obtain a band of collagenolysis at the expected size of MMP-1 (not shown). We detected in all fractions a band corresponding to MMP-2. Moreover, a doublet for MMP-9 likely corresponding to the latent and active forms was found essentially in the cytosolic and membrane fractions. Some of these observations were confirmed by Western blot using anti-MMP-2 and anti-MMP-9 antibodies (Fig. 1D). The ratio between secreted active MMP-2 and active MMP-9 in the supernatant and between membrane-associated active MMP-2 and MMP-9 was quantified. Membrane active MMP-2 was fourfold more important than MMP-9, whereas secreted active MMP-9 was approximately threefold more enriched than active MMP-2. Finally, we detected by Western blot the presence of MT1-MMP in the membrane, nucleus, and cytoskeleton of OE-MSCs, but not in the cytosol fraction (Fig. 1F). Measures of optical densities revealed that the membrane/nucleus and membrane/cytoskeleton ratio for MT1-MMP was 4 and 1.5, respectively. Enrichment of the different fractions was confirmed by Western blot using specific anti-CD44, anti-actin, and anti-histone H3 for the membrane, cytoskeleton, and nuclear fractions, respectively (Fig. 1G).
Two-Dimensional Migration of OE-MSCs and MMPs Immunolabeling Distribution
In an attempt to link the cellular distribution of MMPs and the migratory phenotype of OE-MSCs, we developed a silicone insert assay (Fig. 2). This allowed for rapid compartmentalization of confluent cultured OE-MSCs into two well-defined subpopulations following removal of the silicone insert: (i) a subpopulation of cells at the migration front displaying characteristic highly polarized oyster-like morphology with extensive protrusive activity and large lamellipodia and (ii) a subpopulation of cells laying behind the migration front with a rather symmetric nonpolarized morphology. In these conditions, we performed immunocytochemistry and analyzed the distribution of MMPs combined with F-actin labeling (Fig. 3). We observed clear differences in the pattern of distribution of the different MMPs with regard to their polarity, 24 h after migration onset. Globally, MMP immunoreactivity was heterogeneously distributed in migrating cells, whereas trailing cells displayed a rather homogeneous distribution with occasional clustering in the perinuclear area, as indicated for instance in Figure 3A for MT1-MMP. In migrating cells, MT1-MMP immunostaining showed a clear punctate, vesicular-like pattern, with prominent presence of the protease at the membrane where it colocalized with cortical actin. MT1-MMP was particularly enriched at the edges of lamellipodia all over the migration front. MT1-MMP showed also a perinuclear accumulation (Fig. 3B).
Principle of the silicone stencil assay for synchronized cell polarization/migration experiments OE-MSCs are cultured on a poly(dimethylsiloxane) (PDMS) thin elastic film of defined diameter containing an internal rectangular stencil. When cells reach confluence after 24 h in culture, removing the silicone insert is sufficient to induce cell polarization and trigger cell migration into the empty laminin-coated area of the slide. Cellular distribution of MMP-1, MMP-2, MMP-9, and MT1-MMP in migrating OE-MSCs 24 h after removal of the silicone insert. Representative confocal microphotographs showing the distribution of MT1-MMP in confluent static OE-MSCs (A) and various MMPs in migrating OE-MSCs (B–E) all in green, combined with phalloidin labeling of F-actin (red) and nuclear dye Hoechst #33258 (blue). MT1-MMP immunolabeling in (A) is representative of the distribution observed for all MMPs on static cells behind the migration front. Note that they display a rather homogenous MMP distribution across the cell body with occasional more intense punctate immunolabeling in the perinuclear area (arrow and inset). (B) MT1-MMP distribution in migrating cells. Note MT1-MMP-positive vesicular-like elements streaming from the perinuclear area towards the forefront of the cell (see top inset). MT1-MMP colocalizes (yellow) with cortical actin at the cell membrane in the migration front (see insets). (C) MMP-1 presents a punctate immunolabeling and good colocalization with bundles of actin microfilaments across the cell, including middle and forefront parts of the cell (see arrow and inset). (D) MMP-2 distribution is rather homogeneous across the cell and, in contrast with the other MMPs, not enriched in the front of migration (see inset). (E) MMP-9 is variably colocalized with F-actin across the cell and relatively well colocalized in the migration front (see inset). These observations resulted from the detailed analysis of more than 30 cells per immunostaining in three independent cultures. Scale bars: 10 μm.

MMP-1 displayed a rather punctate distribution, which followed F-actin bundles. MMP-1 colocalized well with F-actin at the centre part of the cell and clustered at some rich compact F-actin spots at the membrane. MMP-1 showed a vesicular-like immunoreactivity homogeneous throughout the cytoplasm, with no particular gradient towards the migration front, as observed for MMP-9 and MT1-MMP (Fig. 3C).
MMP-2 was the most homogeneously distributed among MMPs, exhibiting an immunolabeling profile closely associated with thin F-actin fibers. In contrast, MMP-2 immunostaining was generally negatively correlated with thick bundles of F-actin fibers. Unlike all the other studied MMPs, MMP-2 immunolabeling appeared to be in some cases more intense at the rear of the cell (Fig. 3D).
MMP-9 displayed a more punctate distribution that colocalized well with F-actin, including in some areas of the plasma membrane at the migration front and generally associated with cortical actin. Unlike MMP-2, a good correlation was observed between MMP-9 immunostaining and bundles of F-actin microfilaments. Also distinct from MMP-2, MMP-9 displayed a vesicular-like distribution with a gradient oriented towards the migration front (Fig. 3E).
Further confocal analyses were used to ascertain and quantify colocalization between MMPs and F-actin. Kymographs were constructed and analyzed with ImageJ software from single-pixel width lines taken from each channel of the confocal images. Profiles of the signal intensities of MMPs measured along single-pixel width lines confirmed colocalization of the labeling. Quantification of single plane images in the z-axis of MMP-1, MMP-2, MMP-9, MT1-MMP, and merged puncta revealed a level of colocalization with F-actin as follows: MT1-MMP 72%, MMP-1 69%, MMP-2 64%, and MMP-9 52%.
Two-Dimensional Migration of OE-MSCs and Gelatinolytic Distribution
The high level secretion of MMP-2 and MMP-9 by OE-MSCs raises the question of the role of these proteinases, knowing that OE-MSCs can migrate through long distances in the nervous tissue. MMPs have been involved in the migration of neural cells, including astrocytes (32), OECs (18, 44), and neuroblasts (21, 24). Consequently, we sought to determine the localization of the net proteolytic activity of these enzymes in OE-MSCs using in situ zymography (Fig. 4). In the example for MT1-MMP, Figure 4A illustrates that nonmigrating OE-MSCs showed poor colocalization between MMP inmmunostaining and gelatinolytic activity. On the contrary, migrating OE-MSCs showed gelatinolytic activity distributed along the cell membrane and particularly often at the level of the migration front, notably for MT1-MMP and MMP-9 (Fig. 4B, D). A higher overlap was observed between gelatinolytic activity and immunolabeling for different proteinases outside the migration front, except for MMP-2 (Fig. 4C). It is noteworthy that the MMP-1 antibody did not work when combined with in situ zymography.
In situ zymography on living OE-MSCs 24 h after removal of the silicone insert. Confocal photomicrographs showing in situ zymography in live OE-MSCs revealed with gelatin-quenched fluorescent substrate (green), combined with immunolabeling for MT1-MMP, MMP-2, and MMP-9 (red) and nuclear dye Hoechst #33258 (blue). (A) No significant colocalization between gelatinolytic activity and MT1-MMP was observed in confluent static OE-MSCs behind the migration front. This distribution was representative of that observed for the other studied MMPs in nonmigrating cells (not shown). In migrating cells (B), gelatinolytic activity and MT1-MMP immunostaining colocalized in the perinuclear area and at the membrane in the migration front (see inset). (C) MMP-2 also colocalized with gelatinolytic activity in some discrete areas of the membrane in the migration front (inset) and also occasionally in the perinuclear area. (D) MMP-9 displayed good colocalization with gelatinolytic activity at the membrane in the front of migration (inset) and relatively poor colocalization in the rest of the cell. Scale bar: 10 μm.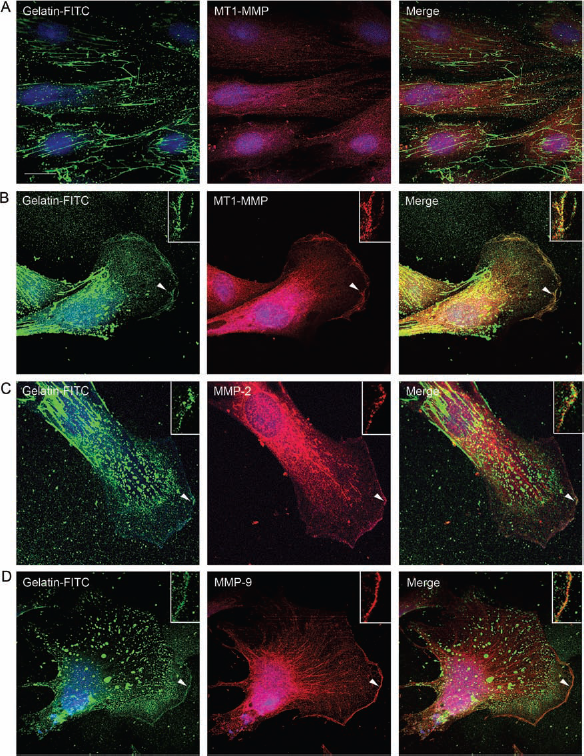
Effects of Selective Inhibitors of MMPs on Three-Dimensional OE-MSCs Migration
We used the Boyden chamber transmigration assay, which may best reflect 3D migration of OE-MSCs in the tissue and the functional implication of MMPs. The role of the latter in OE-MSCs motility was investigated using different modulators of MMP activity (Fig. 5A). In basal medium conditions, around 50% of the total upper side membrane-seeded cells transmigrated through the Boyden filter in 24 h. To discriminate between MMPs and adamalysins (a disintegrin and metalloproteinase, ADAMs), closely related transmembrane metalloproteinases also involved in cell migration (37), we used RXPO3R, a specific inhibitor of MMPs that does not inhibit ADAMs. RXPO3R reduced by 62% the number of transmigrating OE-MSCs in 24 h (Fig. 5B). Next, we used the N-terminal truncated form of TIMP-1 lacking the C-terminal domain, which retains the inhibitory properties of the full length, namely inhibition of secreted MMPs and very poor inhibitory activity against MT-MMPs (19). Truncated TIMP-1 reduced by 42% the transmigration of OE-MSCs, confirming the implication of at least secreted MMPs (Fig. 5B). In all cases, the treatments did not cause any cell damage, as evaluated with the MTT test (data not shown).
Selective MMP inhibitors reduce transmigration of OE-MSCs. (A) Epifluorescence microphotographs of the lower side of Boyden chamber filters showing hoechst-labeled nuclei after transmigration of OE-MSCs. Note that all MMP inhibitors significantly reduce transmigration of OE-MSCs; RXPO3R: broad spectrum MMP inhibitor (1 μM); N-TIMP-1: N-terminal domain of TIMP-1 (3 nM); MMP-2I: selective MMP-2 inhibitor (1 μM); MMP-9I: selective MMP-9 inhibitor (50 μM); LEM-2/15: specific MT1-MMP neutralizing antibody (15 μg/ml). Scale bar: 100 μm. (B) Quantification of OE-MSC transmigration after different treatments. Values represent the means ± SEM of three independent experiments. *p < 0.05, **p < 0.01. ANOVA followed by the Tukey's test.
We asked next whether the different MMPs specifically contributed to transmigration of OE-MSCs. Migrating cell capabilities were assessed using MMP-2 and MMP-9 selective inhibitors and a neutralizing monoclonal antibody against MT1-MMP (LEM2/15) (13). We added MMP-2 and MMP-9 inhibitors and LEM2/15 at 1 μM, 50 μM, and 15 μg/ml, respectively. Each inhibitor consistently inhibited transmigration: MMP-2 inhibitor (38%), MMP-9 inhibitor (63%), and LEM2/15 (50%) (Fig. 5B).
Neurotrophic Effects of OE-MSCs and OECs
As described previously in a mouse model of amnesia, transplanted OE-MSCs migrate to lesioned hippocampus, differentiate into neurons, and promote learning and memory recovery (31). In addition to providing newly formed neurons, grafted OE-MSCs very likely provide trophic support for resident hippocampal stem cells. In order to test this hypothesis, we evaluated the effects of medium conditioned for 72 h by OE-MSCs on neurite outgrowth and axo-dendritic differentiation of cortical neurons. We show that exposure of cortical neurons to OE-MSC-conditioned media for 24 h induces a dramatic 175% increase in the total length of neurites. Furthermore, we observed that conditioned medium from OE-MSCs induces also a 207% increase in the number of neurites exhibiting strong Tau immunolabeling characteristic of ongoing axonal differentiation, with respect to undifferentiated neurites that were immunopositive only for MAP-2 (Fig. 6A, C, D).
Medium conditioned by OE-MSCs induces neurite outgrowth and axo-dendritic differentiation (A) Representative epifluorescence microphotographs of cortical neurons after 24 h in culture exposed to nonconditioned (Ctl) or medium conditioned by OE-MSCs. Neurites were labeled with anti-Tau-1 (green) and anti-microtubule-associated protein-2 (MAP2); (red) specific antibodies to evaluate the degree of axonal and dendritic differentiation, respectively. Note that medium conditioned by OE-MSCs promotes axonal and dendritic outgrowth. Scale bar: 50 μm. (B) Gelatin-based gel zymography showing abundant gelatinase activity in the OE-MSC-conditioned medium (CM) and successful depletion of gelatinases by gelatin-sepharose 4B beads in OE-MSC-depleted medium (DM). (C) Quantification of neurite outgrowth in cortical neurons cultured with nonconditioned medium (Ctl), OE-MSC-conditioned medium (CM), and conditioned gelatinase-depleted medium (DM). (D) Evaluation of axo-dendritic differentiation in the same experimental conditions as (C). Values represent the means ± SEM of at least three independent experiments. *p < 0.05, ANOVA followed by the Tukey's test.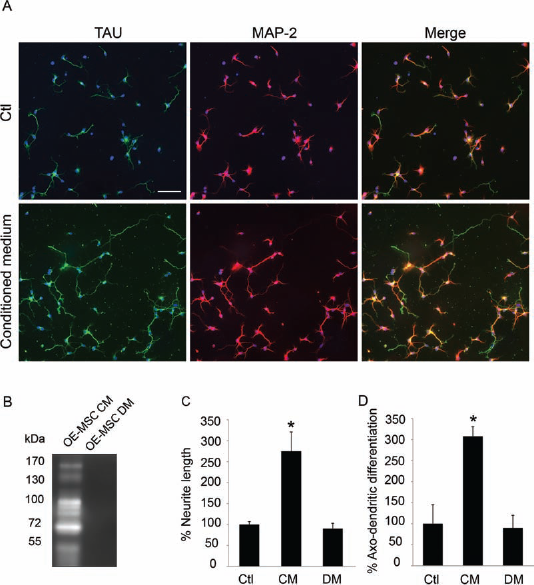
We have also demonstrated in previous studies the ability of OECs to promote in vivo axonal regeneration, remyelination, as well as functional recovery after traumatic injury to the spinal cord (9, 26, 27, 41). However, the underlying mechanisms involved in these therapeutic effects remain elusive. Here we demonstrate that media conditioned by OECs also induce significant increases in neurite outgrowth in both cultured cortical (268%) and DRG (270%) neurons (Fig. 7), indicating that the OECs secrete factors that support neurite outgrowth as well.
Medium conditioned by OECs induces neurite outgrowth in cortical neurons and DRG explants. (A) Epifluorescence microphotographs of cortical neurons treated for 24 h after seeding with nonconditioned medium (Ctl) or with OEC-conditioned medium (CM) and showing neuronal marker anti-βIII tubulin staining. Scale bar: 20 μm. (B) Quantification of neurite outgrowth in cortical neurons treated for 24 h with nonconditioned medium (Ctl), medium conditioned by OECs (CM), and with OEC gelatinase-depleted medium (DM). (C) Epifluorescence microphotographs of dorsal root ganglion (DRG) explants treated for 24 h after explantation with nonconditioned medium (Ctl) or medium conditioned by OE-MSCs (CM) and showing anti-βIII tubulin staining. Scale bar: 100 μm. (D) Quantification of the surface occupied by DRG axons after incubation with different media. Values represent the means ± SEM of at least three independent experiments. *p < 0.01, ANOVA followed by the Tukey's test. Comparisons were made between the CM and DM groups versus the Ctl group and between the CM and DM groups.
MMPs produced by neurons have been previously involved in promoting neurite outgrowth of cortical neurons (16, 33), and we asked next whether gelatinases MMP-2 and MMP-9 secreted by OE-MSCs and OECs could contribute to the paracrine neurotrophic effects of these olfactory cells. Accordingly, we depleted from media conditioned by cells the gelatinases MMP-2 and MMP-9, using gelatin-sepharose 4B beads that sequester the enzymes. This method was originally described as an efficient way to concentrate MMP-2 and MMP-9 in tissue extracts with low content on these enzymes (47). The incubation of cortical neurons with gelatinase-depleted media resulted in the abrogation of the neurotrophic effects induced by the conditioned media from OE-MSCs, and no differences were observed in global neurite length and axo-dendritic differentiation with untreated controls (see Fig. 6).
Depletion of gelatinases from OEC-conditioned medium did not provoke a complete abolition of its trophic effects on cortical neurons but caused nevertheless a strong inhibition (Fig. 7B). The same depletion abrogated the trophic effects on DRG axons (Fig. 7D).
Discussion
The nasal olfactory mucosa is a source of cells for transplantation and repair of the nervous system. Successful cell therapy in the CNS requires transmigration of grafted cells to their target areas across different tissues, including the brain parenchyma and the basal lamina of brain blood vessels. Moreover, transplanted olfactory cells establish a functional interplay with the microenvironment of the host tissue, setting the conditions for their phenotypic differentiation and influencing the biology of surrounding endogenous cells. Most mechanisms of action governing these phenomena remain elusive, and gaining insight into these mechanisms is important to improve the therapeutic potential of grafted cells, in particular OE-MSCs and OECs. The present work is in keeping with this idea and provides the first evidence that (1) several MMPs are readily expressed in OE-MSCs cultured in the same conditions as those used for grafted cells; (2) the distribution of MMPs and their proteolytic activity in OE-MSCs change when they adopt the polarized phenotype characteristic of migratory cells; (3) several MMPs contribute to OE-MSC migration, as demonstrated in an in vitro 3D transmigration test; (4) OE-MSCs, as demonstrated previously for OECs, secrete neurotrophic factors that promote neuronal growth and differentiation; and (5) MMPs may be part of the factors secreted by olfactory mucosa cells that display neurotrophic activities on CNS and PNS neurons.
MMPs Are Readily Expressed in OE-MSCs
A prerequisite for the successful use of OE-MSCs in cell therapy is that they remain endowed with the molecular machinery that allows them to penetrate into tissues and bypass barriers, including basal lamina from blood vessels and normal and scarred brain parenchyma. The MMPs are in principle good molecular candidates of such machinery because of their ability to cleave/degrade most ECM protein components. Neural precursors in culture express several MMPs, including MMP-2 or MMP-9, and the four TIMPs (1, 11). In vivo, cerebellar progenitors also abundantly express MMPs when they migrate from the inner to the outer molecular layer during mouse postnatal development (44), and MMP-9 is particularly upregulated in endogenous progenitor cells that migrate from the SVZ into lesioned areas (i.e., striatum) following brain ischemia (24). In agreement with previous reports, the present findings reinforce the idea that MMPs are widely distributed across stem cell populations, stressing their functional importance. It is interesting to note that among gelatinases, the pro- and active forms of MMP-9 are found in the supernatant and cytosol principally. Both forms are also detected in the membrane by highly sensitive gelatin zymography approaches. Instead, the MMP-2 pro-form is essentially secreted, whereas the active form appears to be preponderant in the cytosol and membrane compartments. Since gelatin gel zymography is not sensitive enough to detect MMP-1, we ran collagen zymography gels that offer a higher sensitivity for this proteinase. Accordingly, we detected a band of collagenolysis at 55 kDa, compatible with the expected molecular size of MMP-1 and in agreement with previous findings in other cell types (12). Finally, using specific anti-MT1-MMP antibodies and Western blot, we detected an immunoreactive band at the expected molecular size (60 kDa) in the membrane, nucleus, and cytoskeleton fractions. Very weak immunoreactivity was found in the cytosol fraction, indicating that the pool of intracellular MT1-MMP is mainly associated with membrane, cytoskeletal, and nuclear elements.
Polarization of OE-MSCs Upon Migration Influences MMPs Distribution and Proteolytic Activity
We used the stencil method of 2D migration, previously described to analyze collective migration of Madin-Darby canine kidney (MDCK) cell lines (36), with some modifications to optimise its application to OE-MSCs. The method allows for synchronized migration and polarization of cells in the migration front while keeping trailing cells in a packed, rather immobile configuration. Upon removal of the silicone stencil OE-MSCs migrate into the laminin-coated territory within a few hours. Using immunocytochemistry for MMP-1, -2, -9 and MT1-MMP, we observed that in most cases the MMPs adopted a polarized distribution that corresponded for the most part to the phenotype displayed by the actin cytoskeleton, as previously reported in neural cell types (32, 40). Nevertheless, the distribution of MMPs displayed some specificities: MMP-2 appeared to be more homogeneously distributed, whereas MMP-1, MMP-9, and MT1-MMP were more enriched in the migration front associated with cortical actin and also with bundles of actin microfilaments across the cell in the case of MMP-1. Overall, the level of colocalization with the actin cytoskeleton was comparable across MMPs, being in all cases over 50%. MMP-1, MMP-9, and MT1-MMP clearly presented a more punctate immunoreactivity than MMP-2; the latter often localized between the perinuclear area and the front of migration, aligned with the actin cytoskeleton. This may be reminiscent of vesicular transport from the trans-Golgi network to the membrane (40, 45, 49).
The distribution of MMPs partially overlapped with the gelatinolytic activity revealed by in situ zymography. Gelatinolytic activity represents the net balance between the proteolytic activity of gelatinases and their inhibition by their endogenous inhibitors. Among MMPs, MMP-2, MMP-9, and MT1-MMP have a good affinity for gelatin. Accordingly, some colocalization was observed when combining in situ zymography with immunocytochemistry for these proteinases. Most interestingly, the main colocalization was found in the migration front, especially for MT1-MMP, suggesting the presence of a pool of active proteinases in this functional and motile area. In contrast, trailing cells exhibited a rather homogenous gelatinolytic activity across the cell in phase with a nonpolarized phenotype and the absence of motility.
MMPs Are Involved in the Transmigration of OE-MSCs
A key point of the present work is that data on MMP distribution and polarization of the migrating cells correlate with data on the role of these MMPs in migration. We used this time a 3D in vitro migration system, which may more closely reflect transmigration of grafted cells across tissues. In these conditions, all the tested modulators of MMP activity were efficient in inhibiting OE-MSCs transmigration in Boyden chambers. RXPO3R, known to inhibit MMPs and MT-MMPs but to spare ADAMs (5), was very efficient, showing over 60% inhibition of migration. This inhibitor has been successfully used previously to demonstrate inhibition on neurite outgrowth and hence the implication of MMPs in this process (33). Also, the specific MMP-9 inhibitor reached over 60% efficiency in inhibiting migration, stressing the relative importance of this proteinase among MMPs in cell migration, as it is also the case during neural precursor cell migration in vivo (24, 44). A lower level of inhibition was obtained with the N-terminal domain of TIMP-1 and the MMP-2-specific inhibitor. The latter caused less than 40% inhibition. This is in contrast with the nearly abolition of OEC migration obtained with the same concentrations of the MMP-2 inhibitor (18). Two major conclusions can be drawn from the comparison between these studies that indicate overall a relative cell-dependent specificity in the role of MMPs in nasal olfactory cells migration: (i) OEC migration may essentially require MMPs, whereas proteinases from other families may also contribute to the motility of OE-MSCs, and (ii) among gelatinases, MMP-2 seems to play a more prominent role than MMP-9 in OECs, whereas MMP-9 displays a greater effect in OE-MSCs.
Do MMPs Contribute to the Neurotrophic Effects of OE-MSCs and OECs?
The traditional view of stem cells as mere replacement for damaged cells is broadening along with new therapeutic prospectives, including the neurotrophic effects of undifferentiated grafted cells in host tissues. Our data reveal for the first time that this is the case for OE-MSCs. Indeed, the conditioned media of OE-MSCs induces a dramatic increase in the length of neurites from immature cortical neurons in a short period of time (24 h). Noticeably, the factors released by olfactory cells promote also rapid axo-dendritic differentiation of these neurons. Even stronger neurotrophic effects were obtained upon incubation of cortical neurons with conditioned media from OECs. Moreover, OECs also induce striking growth of explanted axotomized DRG axons, pointing out altogether high potential for nerve repair in both CNS and PNS. Other studies have shown that transplantation of CNS progenitor cells into explants of degenerating retina promotes neurite extension in response to increased MMP-2 secretion by host glial cells (48). Similarly, coculturing adult retinal cells and rat olfactory ensheathing cells results in axon growth mediated by MMP-2 produced by the latter (35). In an attempt to further assess the possible neurotrophic contribution of secreted MMPs, we used gelatin-coated beads to deplete the MMP-2 and MMP-9 gelatinases from media conditioned by olfactory cells. This resulted in abrogation of OE-MSCs and OECs neurotrophic properties, reinforcing the idea of a paracrine neurotrophic contribution of these MMPs that adds to previously reported autocrine effects (16, 17, 33). The neurotrophic effects of MMP-2 and MMP-9 may be related to their capacity to cleave extracellular matrix components that facilitate the progression of neurites across their microenvironment. Alternatively and not exclusively, MMPs may exert their neurotrophic effects by influencing the activity of neurotrophic factors. It is now established that some MMPs (e.g., MMP-7 and MMP-3) may proteolytically process the inactive immature forms of BDNF and NGF into their biologically active forms (23).
Taken together, these findings highlight positive roles that endogenous MMPs from OE-MSCs and OECs may play in the therapeutic properties of these cells via the control of their migratory and neurotrophic capabilities. These data may be relevant to optimize the therapeutic potential of grafted OE-MSCs and OECs and advise caution on the indiscriminate use of metalloproteinase inhibitors in the prospects of repair and regenerative strategies in the nervous system.
Footnotes
Acknowledgments
This work was supported by funding to the UMR 7259 laboratory from the CNRS and the Aix-Marseille University and by grants to M.K., S.R., and F. F. from the Institut pour la Recherche sur la Moelle Epinière (IRME) and from the Direction Générale de l'Armement (DGA). The work was also supported by grants to S.R. and M.K. from the Association Française contre les Myopathies (AFM), the ANR (TIMPAD 08-MNPS-042), and the COST Action BM1001 “Brain Extracellular Matrix in Health and Disease”. We acknowledge support from “Fonds Européen de Développement Régional” FEDER in PACA. A.O.-Y was recipient of a doctoral fellowship from the French Ministry of Research and from the AFM. O.S. was recipient of a doctoral fellowship from the Association Bir el Bey (Tunisia) and from AFM. Y.G. was recipient of a doctoral fellowship from the Letten Foundation. We thank Dr. Hideaki Nagase (Imperial College London, UK) for kindly providing the N-terminal domain of TIMP-1. We also thank Dr. Alicia Garcia Arroyo (CNIC, Madrid, Spain) for generously providing the LEM-2/15 anti-MT1-MMP antibody. The authors declare no conflicts of interest.