
Abstract
Transplanted olfactory ensheathing cells (OECs) contribute to functional recovery in a range of CNS injuries by several mechanisms, one of which is potentially their ability to form myelin sheaths. OECs sourced from donors of different ages have been shown to remyelinate in several in vitro and in vivo models. However, the optimal donor age for OEC associated remyelination is unclear. This project directly compared the remyelinating potential of p75 purified OEC transplants from three donor ages. OECs were sourced from the olfactory bulbs of embryonic, neonatal, and adult rats and purified by immunopanning, and their remyelinating potential was directly compared by transplantation into the same adult rat toxin-induced model of spinal cord demyelination. Remyelination efficiency 3 weeks after transplantation was assessed morphologically and by immunostaining. Our results indicate that all donor ages remyelinate; however, this process is most efficiently achieved by embryonic-derived OECs.
Introduction
Olfactory ensheathing cells (OECs), the specialized glia of the peripheral olfactory system, have been reported to have diverse regenerative effects when transplanted into a range of CNS pathologies (25,28,31). One such property is their ability to form myelin sheaths around axons of appropriate diameter, a regenerative process especially relevant to conditions featuring primary demyelination such as traumatic spinal cord injury or, more especially, demyelinating diseases such as multiple sclerosis (MS) and leukodystrophies (13).
OECs can be grown in tissue culture from different aged donors—embryonic, neonatal, and adult—as well as from different regions of the peripheral olfactory system (olfactory epithelium and nerve fiber layer on olfactory bulb) (14). Most transplantation studies demonstrating remyelination by transplanted OECs have involved the use of either neonatal or adult-derived cells (2,12,18,19). However, the comparative myelinating capacities of OECs from donors of different ages, information pertinent to optimizing their effectiveness in therapeutic transplantation, have not been assessed in a direct head-to-head comparison using the same lesion model. That age-associated differences exist is suggested by in vitro studies that demonstrate a greater myelination propensity of embryonic-derived OECs (eOECs) compared to adult-derived OECs (aOECs) (10,24), which is in line with other myelinating cell types such as the oligodendrocyte lineage (30,34).
In this study, we test the myelinating capacity of eOECs, neonatal-derived OECs (nOECs), and aOECs using a well-established model of focal spinal cord demyelination (Fig. 1). This model involves induction of a lesion that predominantly consists of primary demyelination by the direct injection of ethidium bromide (EB). The endogenous remyelination that normally follows EB injection is suppressed by locally exposing the area of spinal cord in which the lesion is induced to a high dose of X-irradiation (EB-X lesion model) (5). Given the debate that has occurred around the origin of peripheral-type remyelination observed following OEC transplantation in this model (6), we have used lentiviral vectors to genetically and unequivocally label cells prior to transplantation with green fluorescent protein (GFP).
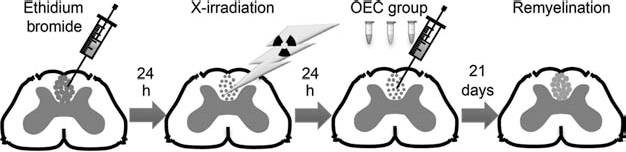
Experimental design. Demyelination of the dorsal funiculus of the rat spinal cord was induced by direct injection of ethidium bromide (EB). Spontaneous remyelination was abolished by X-irradiating the demyelinated area 24 h following EB injection. On the next day, 50,000 embryonic (e), neonatal (n), or adult (a) purified olfactory ensheathing cells (OECs) were injected into the center of the dorsal funiculus using the same laminectomy prepared for toxin injection. Remyelination was assessed 21 days following transplant.
Materials and Methods
Cell Culture
OECs were obtained from olfactory bulbs (OB) of Fischer 344 rats (Harlan, Shardlow, Derbyshire, UK) from embryonic day 18 fetuses (eOEC), 7-day-old pups (nOEC), and 12-week-old female adults (aOEC) as previously described (3,10,24). Once removed, OEC-containing regions of OBs were enzymatically and mechanically dissociated in a modification of a previously detailed protocol (24). Briefly, residual meningeal tissue and blood vessels were carefully stripped from all extracted OBs. Whole embryonic OBs and the rostral third of neonatal and adult OBs (containing the nerve fiber layer) were chopped with a scalpel blade and then enzymatically digested using 0.125–0.25% Trypsin (Worthington Biochemical Corporation, Lakewood, NJ, USA) and 50 μg/ml DNase I (Sigma-Aldrich, Dorset, England, UK) in Ca/Mg-free Hank's buffered saline solution (HBSS; Invitrogen, Paisley, Scotland, UK) for 25–60 min at 37°C. Following addition of serum-containing medium [DF10S; composed of Dulbecco's modification of Eagle's medium (DMEM; Invitrogen) and Ham's F12 medium (Invitrogen) at a 1:1 ratio (v/v), 10% fetal bovine serum (FBS; v/v; Biosera, Uckfield, East Sussex, UK), 2 mM l-glutamine (Invitrogen), and 50 μM gentamicin (Invitrogen, UK)], the tissue was mechanically dissociated by gentle trituration 10–20 times each through two progressively narrower fire-polished glass pipettes to create a single cell suspension. The homogenate was centrifuged at 300×g for 5 min and resuspended in DF10S with mitogens [DF10S+mit; composed of DF10S supplemented with 20 μg/ml bovine pituitary extract (Invitrogen) and 2 μM forskolin (Sigma)]. Cells were plated on poly-l-lysine (PLL)-coated 100-mm Petri dishes (Corning, Corning, NY, USA) and incubated at 37°C with 5% CO2. Fresh media were added after 4 days and every 2–3 days thereafter until purification.
Purification by p75NTR Immunopanning
To compare highly pure preparations of OEC cultures, mixed OB cultures were immunopanned to enrich for p75 neurotrophin receptor 192 IgG (p75NTR)-expressing OECs in the cultures (24). Briefly, immunopanning was carried out 5, 7, and 10 days after plating for eOECs, nOECs, and aOECs, respectively. One hundred millimeter nontissue culture-treated Petri dishes (Corning) were incubated overnight at 4°C with goat anti-mouse IgG, IgA, and IgM antibody (Cappel, Cochranville, PA, USA) diluted 1:100 in 0.05 M Tris buffer (pH 9.5; Sigma). Dishes were rinsed thoroughly with L-15 before a proportion was incubated with monoclonal anti-p75NTR 192 IgG antibody (mouse monoclonal IgG from hybridoma cell line 192 supernatant; Dr. G. Plant) diluted 1:10 in L-15 for 2 h at 4°C. Dishes were washed again thoroughly with L-15 before use. Cell suspensions of the unpurified cultures were first incubated with goat anti-mouse antibody-coated dishes to remove nonspecific binding cells, such as microglia for 30 min at room temperature (RT). The supernatant was then incubated with the p75NTR-coated dishes for 30 min at 4°C to collect OECs. Dishes were then washed vigorously with L-15 to remove loose and unbound cells. Adherent cells were fed with DF10S+mit and incubated at 37°C with 5% CO2 for 2 days before replating 1:1 onto PLL-coated dishes and left to increase in number for transplant. When cultures became confluent, they were passaged at 1:2 for a maximum of two passages (13– 29 days following initial OB extraction from donors).
Immunocytochemistry
At each passage and on the day of transplant, aliquots of OEC cultures were taken and plated upon PLL-coated 13-mm glass coverslips and cultured for 24 h in DF10S+mit. Live cultures were rinsed three times with L-15 medium before incubation with primary antibodies against p75NTR (1:10, mouse monoclonal from hybridoma cell line 192 supernatant; Dr. G. Plant) or Thy1.1 (1:5, mouse monoclonal from hybridoma cell line supernatant; Dr. G. Plant) diluted in L-15 with 10% normal goat serum (NGS, Sigma) for 30 min at 4°C, before 3 L-15 rinses and incubation with Alexa 555 goat anti-mouse secondary antibody (Invitrogen) for 30 min at 4°C. After three further L-15 rinses, cells were fixed with 4% paraformaldehyde (PFA; Sigma) in PBS for 15 at RT and permeabilized with PBS containing 4% PFA and 0.2% Triton X100 (Sigma) for 10 min at RT. Cultures were rinsed three further times with PBS to prepare them for further staining. Primary antibodies against p75NTR (Promega, Madison, WI, USA), S100b (Dako, Carpinteria, CA, USA), glial fibrillary acidic protein (GFAP; Dako), fibronectin (Dako), calponin (Abcam, Cambridge, Cambridgeshire, UK), laminin (Sigma), nestin (Abcam), and polysialylated neuronal cell adhesion molecule (PSA-NCAM; ABC Scientific, Pasadena, CA, USA) were diluted in PBS containing 10% NGS with 0.2% Triton X100 (diluent) and applied to coverslips for 45 min at RT. After three further PBS washes, coverslips were incubated with Alexa 488 secondary antibodies (Invitrogen) diluted 1:500 in diluent for 30 min at 4°C. A final series of three washes with PBS and one wash with deionized water before mounting coverslips cell-side down on microscope slides using Vectashield containing DAPI (Vector Laboratories, Inc., Burlingame, CA, USA) and sealed using nail varnish. Immunofluorescent images were taken using a Zeiss microscope (Cambridge, Cambridgeshire, UK). Percentage purity was calculated by counts of antibody combination staining from images containing at least 300 cells per field.
Lentiviral Production, Concentration, Titration, and Transduction
OECs were transduced with a replication-incompetent lentiviral vector (pCDH-EF1-copGFP) containing cope-pod GFP with the constitutively expressed cellular polypeptide chain elongation factor 1α (EF1). The lentiviral vector was produced by cotransfection of the transfer vector (pCDH-CMV-MCS-EF1-copGFP; SBI System Biosciences, Mountain View, CA, USA) and Virapower™ packaging mix (Invitrogen) of HEK 293T cells (ATCC, Teddington, Middlesex, UK). At 24 and 48 h after transfection, 45-μm filtered supernatants were collected, pooled, and concentrated in a modification of Coleman and colleagues (9). Briefly, 30-ml aliquots of viral supernatants were carefully loaded onto a 220-μl cushion of 60% iodixanol (from the Optiprep solution; AxisShield, Dundee, Scotland, UK) in a 30-ml conical-bottommed centrifuge tube (Beckman Coulter, High Wycombe, Buckinghamshire, UK) and centrifuged at 50,000 g for 2.5 h at 4°C using a Beckman SW28 swinging bucket rotor. The supernatant from each tube was carefully removed to within 5 mm of the media–iodixanol interface. The remainders were combined and shaken at 200 rpm at 4°C for 2 h before centrifuging in a 3-ml conical bottommed tube (Beckman) at 6,100×g for 22 h at 4°C using a Beckman SW50.1 swinging bucket rotor. The supernatant was discarded, and the pellet was carefully resuspended in 100-μl ice-cold sterile PBS and stored in 10-μl aliquots at −80°C.
Following concentration, the volume of the viral stock required to infect all OECs for transplant was determined. The infectivity of the lentivirus in HEK 293T cells was determined by seeding 5×104 cells per well in DF10S on a 24-well plate with limiting dilutions of the viral stock (1, 0.1, and 0.01 μl) in the presence of polybrene (5 μg/ml; Sigma). After 24 h, the medium was refreshed, and 48 h following transfection, the total number of GFP+ cells was estimated using a hemocytometer and fluorescent microscope (10×, Zeiss) to determine the number of transducing units (TU; infectious particles) per milliliter of viral stock (TU/ml). The number of infectious particles needed to infect one HEK 293T cell was calculated and used to determine multiplicity of infection (MOI) for OECs. MOI was determined using a modification of the protocol used by Ruitenberg and colleagues (27). Briefly, 6,000 OECs per well were seeded onto a 12-well plate and fed overnight with DF10s with 20 μg/ml bovine pituitary extract (BPE; Invitrogen) and 2 μM forskolin (Sigma). The medium was then changed to DF10s with low mitogens, containing 2 μg/ml BPE and 0.2 μM forskolin. Increasing MOI (1, 10, 25, 50, and 100) was added, and then the medium was changed for DF10S+mit after 24 h. After a further 48 h, total GFP+ OECs were estimated as above. The volume of viral stock needed to infect 100% of OECs was taken as the lowest MOI with 6,000 OECs infected. A subset of OECs from each age was infected with the lentivirus according to the protocol of Ruitenberg and colleagues (27). Briefly, 48 h before transplant, 106 cells from each age group were seeded onto 60-mm PLL-coated tissue culture dishes and fed overnight with DF10s with low mitogens. This was refreshed the following day with media containing 5×107 infectious particles (33 μl viral stock, MOI = 50), and 24 h later, the cells were washed with PBS and prepared as normal for transplant (see below). An aliquot was taken from each age and cultured in parallel with the in vivo experiment for check stability of transfection and purity. Transduction efficiency of virus-treated cultures was determined from aliquots of prepared cells on the day of transplantation by counting labeled and unlabeled cells in 10 randomly selected fields using an epifluorescent microscope (Zeiss). Second counts were made at the conclusion of the corresponding in vivo experiment.
Preparation of Cell Cultures for Transplantation
Cells for transplantation were harvested from culture dishes by rinsing twice with prewarmed HBSS followed by exposure to 0.05% trypsin and 0.02% EDTA (Invitrogen) in HBSS at 37°C for 3 min, after which enzyme activity was blocked by the addition of an equal volume of DF10S. The cell suspension was centrifuged, resuspended in 1 ml MEM-HEPES (Invitrogen), and transferred to a 1.5-ml microcentrifuge tube (Starlab, Milton Keynes, Buckinghamshire, UK), and the total number of cells was determined using a hemocytometer. After a further centrifugation, the cells were resuspended in an appropriate volume of MEM-HEPES to produce a final concentration of 50,000 cells/μl and stored on ice prior to injection. An aliquot of cells was taken for final purity determination as outlined above.
Animal Groups
Adult (12–16 weeks) female rats of the inbred, syngeneic Fischer 344 strain were used for all surgical procedures. In total, 41 animals had surgical procedures, 35 of which were transplanted with OECs from embryonic (n = 12), neonatal (n = 11), or adult (n = 12) donors. In each group, four animals were transplanted with GFP-labeled OECs. The remaining six animals were sham-transplanted with MEM-HEPES only.
Surgical Procedures
All surgical procedures were performed in accordance with Home Office UK requirements and National Health and Medical Research Council of Australia. Under anesthesia induced and maintained by isofluorane (Abbott Laboratories, Abbott Park, IL, USA), a dorsal laminectomy was performed on the T13 vertebra to expose the dorsal aspect of the spinal cord. A slit in the dura mater was made with a dental needle, and with the aid of a three-way micromanipulator, the tip of a glass micropipette attached to a 10-μl Hamilton syringe (Hamilton Company, Reno, NV, USA) was positioned 1-mm ventral to the surface of the cord within the dorsal funiculus. Then, 1 μl of a 0.1% solution of EB (in 0.9% saline, v/v) was injected over 1 min to create a discrete area of demyelination. Directly over the lesion site, the lumbar muscles were sutured before the skin was closed with 5–0 polyamide sutures (Ethilon, Ethicon, San Angelo, TX, USA). To suppress endogenous remyelination by X-irradiation, rats were restrained by intramuscular injection of a fluanisone and fentanyl mixture (Hypnorm, 0.30 ml/kg; Janssen Pharmaceuticals, Titusville, NJ, USA) and a 4-cm length of spinal cord centered on the T11 and including the lesion was exposed to 40 grays of X-irradiation using a 255-kV orthovoltage radiotherapy machine (Pantak, Reading, Berkshire, UK). Transplants occurred 24–48 h after irradiation. A similar surgical method to that used for lesion induction was used to inject 1 μl of either this cell suspension or sterile MEM-HEPES only (for controls) into the center of the lesion as identified by the slit in the dura. Perioperative analgesia (Buprenorphine 0.01 mg/kg, SC; Vetergesic, Alstoe Limited, Animal Health, Sheriff Hutton, Yorkshire, UK) and antibiotics (Depocillin, SC; Intervet, Bendigo East, Victoria, Australia) were given to every animal with each surgical procedure.
Light and Electron Microscope Analysis of OEC Remyelination
Three weeks after transplant, seven to eight animals in each transplant group and four in the control group were deeply anaesthetized with pentobarbitone (Animal Care Ltd., Dunnington, York, UK) before transcardial perfusion with 4% glutaraldehyde (Sigma) in PBS. A 1-cm length of spinal cord containing the lesion was cut into 1-mm coronal blocks and processed to retain their craniocaudal orientation. Blocks were osmicated, dehydrated through a series of ethanols, and embedded in TAAB resin (TAAB Laboratories, Aldermaston, Berkshire, UK). Sections of 1 μm were stained with toluidine blue for light microscopy to assess extent of characteristic peripheral-type remyelination by OECs by light microscopy. Ultrathin sections from selected blocks were cut and stained with uranyl acetate and lead citrate (both from Merck, Hoddesdon, Hertfordshire, UK) for examination using a Hitachi H600 transmission electron microscope (Hitachi High Technologies, Maidenhead, Berkshire, UK) to assess OEC remyelination quality.
Immunohistochemistry
The remaining animals in each group were deeply anesthetized using pentobarbitone and transcardially perfused with ice-cold 4% PFA (Sigma) in PBS before spinal cords were prepared for cryosectioning. Sections of 12-μm thicknesses were serially thaw-mounted before immunoassaying. Slides containing lesioned sections were stained with peripheral myelin protein zero (P0) to determine whether GFP-labeled cells colocalize with axon-enwrapping peripheral myelin. Slides were permeabilized with 0.3% Triton X100 in PBS for 15 min at RT, blocked in PBS containing 10% BlokHen (Aves Labs, Tigard, OR, USA) for 45 min at RT, before overnight incubation at 4°C in a humidified chamber with an anti-P0 antibody (1:500, Aves Labs, in PBS with 1% BlokHen). This was followed by a 1-h incubation at RT with a biotinylated goat anti-chicken secondary antibody (1:500, Aves Labs), then a 1-h incubation at RT with a streptavidin-coupled Alexafluor 568 antibody (1:1,000, Invitrogen, in PBS with 1% BlokHen) in darkness. Between each incubation step, slides were washed twice in PBS. Coverslips were then mounted on slides using Vectashield containing DAPI (1.5 μg/ml, Vector Laboratories, Inc.) and viewed by epifluorescent (Zeiss) and confocal (Leica, Milton Keynes, Buckinghamshire, UK) microscopy.
Remyelination Quantification, Scoring, and Statistical Analysis
The proportion of P0+ myelin sheaths associated with GFP-labeled OECs was determined by analyzing the central transverse section from the lesions of three animals in each age group. Myelinating OECs were identified by GFP-labeling, usually colocalizing with a DAPI-stained nucleus, in direct apposition with a P0-immunostained peripheral myelin ring. The presence of a P0-immunostained ring without accompanying GFP indicated either an unlabeled OEC or an endogenous Schwann cell.
The presence and extent of peripheral “signet ring” appearing myelination by OECs (12) was assessed using a remyelination scoring system (17,22). Briefly, two researchers (D.J.C.C. and C.E.H.), blinded to the transplant paradigm, ranked each slide in order of remyelination completeness using toluidine blue-stained resin sections. Statistical analysis was performed using Graphpad Prism (San Diego, CA, USA) and Microsoft Excel software (Redmond, WA, USA). Ranking data were analyzed using a nonparametric Kruskal–Wallis test followed by a Dunn's post hoc test. Other differences among control and experimental groups were analyzed by t test or one-way ANOVA. Differences were considered significant when p ≤ 0.05.
Results
OEC Preparations of High Purity Were Obtained From Embryonic, Neonatal, and Adult Olfactory Bulb
Similar purity was required to distinguish the effect of age on OEC myelinating ability, as different levels of contaminating cells can alter the myelinating properties of OECs (21). We first obtained OEC populations of high purity by immunopanning against the 192 IgG low-affinity neurotrophin receptor (p75NTR), an approach routinely used by several groups to isolate neonatal and adult OECs (21,24,29). Cultures were passaged twice after immunopanning to increase cell number for transplantation, with embryonic cultures becoming confluent in roughly half the period of neonatal and adult cultures (Table 1). Cultured cells were characterized by immunostaining at each passage to confirm OEC identity and purity and to determine the number and identity of contaminating cell types (Fig. 2). OECs were identified by expression of p75NTR and either S100 or diffuse, low level GFAP (Fig. 2A–F). OECs also expressed laminin and nestin, and NCAM at sites of contact (data not shown). OECs from all ages displayed several morphologies ranging from spindle shaped to process bearing or flat (8). At least 98% of cells were identified as OECs by p75NTR expression in all cultures immediately after immunopanning (Table 1). OEC purity remained very high over two subsequent passages across the different ages with at least 93.5% of cells expressing p75NTR. Cells that were p75NTR negative were very scarce after immunopanning and only marginally increased with passage (Table 1). These were identified as mainly fibroblast and/or meningeal cells by their morphology and either fibronectin, Thy1.1, or calponin expression, consistent with previous studies (16,33) (Fig. 2G–P). OECs did not express calponin or PSA-NCAM (data not shown). Very few p75NTR-negative, strongly GFAP-positive cells with a fibrous morphology resembling astrocytes were observed (Fig. 2E). The purity of OECs in an aliquot of cells taken from the transplant preparation on the day of transplantation was determined to be, on average, 97.53 ± 2.14%, 98.91 ± 0.29%, and 96.9 ± 0.40% for embryonic, neonatal, and adult OEC transplants, respectively (Table 2). Thus, OEC transplants from each age were highly pure, and importantly, this purity was not significantly different between groups (one-way ANOVA, p = 0.55).

The antigenic profile of cultured OECs from embryonic (A, D, G, K, N), neonatal (B, E, H, L, O), and adult donors (C, F, I, M, P). OECs displayed highly similar morphological and antigenic properties at each age. OECs expressed p75 in combination with either S100 (A–C) or glial fibrillary acidic protein (GFAP) (D–F). Very occasionally, a strongly GFAP-positive cells characteristic of astrocytes were seen (E, arrow). Small numbers of fibronectin-expressing (G–I), thy1.1-expressing (K–M), or calponin-expressing (N–P) cells with morphologies of fibroblasts or meningeal cells were rarely observed (G, H, K, O, P, arrows). Scale bar: 100 μm.
Antigenic Profile of Immunopanned eOEC, nOEC, and aOEC Cultures Over Time In Vitro
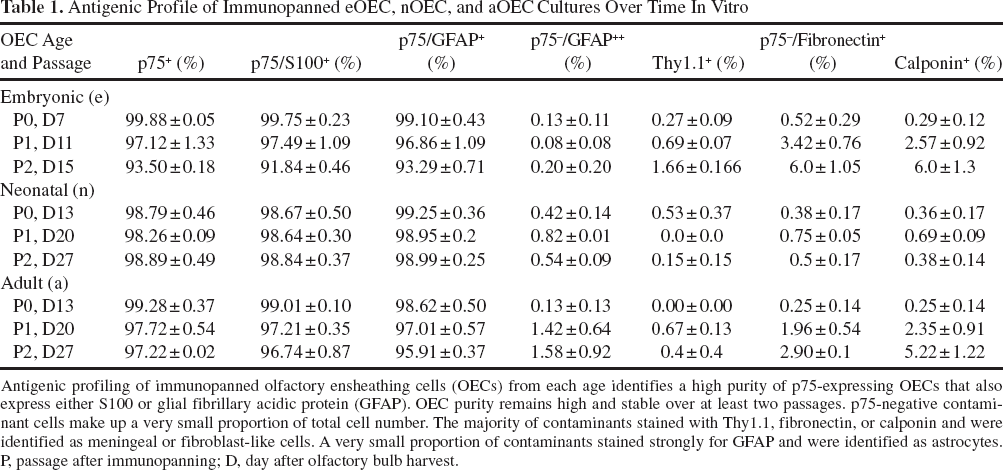
Antigenic profiling of immunopanned olfactory ensheathing cells (OECs) from each age identifies a high purity of p75-expressing OECs that also express either S100 or glial fibrillary acidic protein (GFAP). OEC purity remains high and stable over at least two passages. p75-negative contaminant cells make up a very small proportion of total cell number. The majority of contaminants stained with Thy1.1, fibronectin, or calponin and were identified as meningeal or fibroblast-like cells. A very small proportion of contaminants stained strongly for GFAP and were identified as astrocytes. P, passage after immunopanning; D, day after olfactory bulb harvest.
Purity of Transplanted eOEC, nOEC, and aOEC Cultures

An aliquot of the transplant preparation was taken for purity determination at time of transplant. Each batch used for transplant was highly pure, and there was no significant difference in purity between groups (one-way ANOVA, p = 0.55).
Transplants From Each Donor Age Result in OEC-Like Remyelination of the Demyelinated Spinal Cord
To establish whether different donor ages of OECs affected their ability to remyelinate areas of CNS demyelination, purified preparations of embryonic, neonatal, or adult OECs were transplanted into the X-irradiated, ethidium bromide-demyelinated dorsal funiculus of the spinal cord of adult syngeneic rats (Fig. 2). The extent of remyelination was compared 3 weeks following transplantation. Light microscopy of toluidine blue-stained semithin resin sections of lesions from media-only control animals showed that no remyelination of any kind was observed (Fig. 3A, B). Control lesions were dominated by dark staining myelin debris-filled macrophages and demyelinated axons (Fig. 3B, inset). Light microscopy of toluidine blue-stained semithin resin sections of lesions from each donor age showed extensive remyelination by peripheral type myelin characteristic of OEC transplants (12) (Fig. 3C-H). Lesions were identified by their lighter overall staining and the presence of dark, myelin-filled macrophages. Remyelination by peripheral type myelin was identified by a dark-stained myelin sheath around axons and surrounded by a layer of cytoplasm of variable thickness, often containing the nucleus. This arrangement creates a “signet ring” appearance in cross-section that is relatively easy to distinguish from the central type myelin (Fig. 3D, F, H insets). No central-type remyelination was observed within lesions in any of the experimental groups.
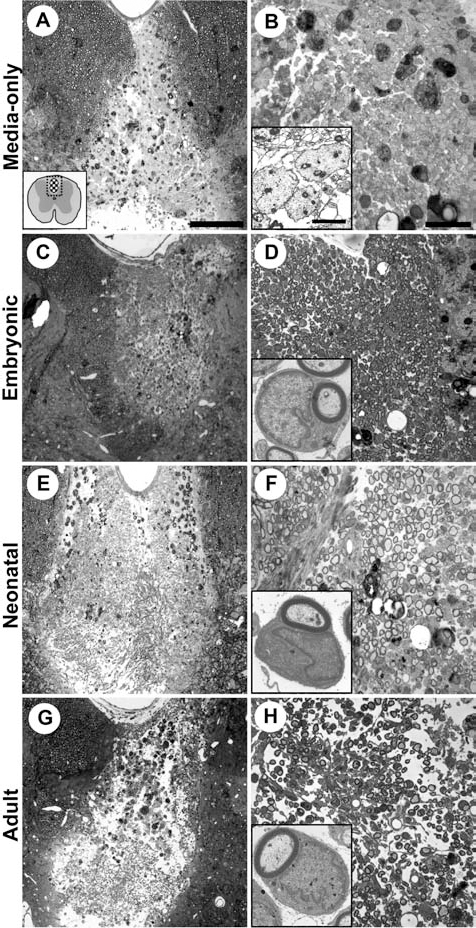
Embryonic, neonatal, and adult OECs myelinate the demyelinated spinal cord. Resin sections through the dorsal funiculus of control and OEC-transplanted rat spinal cord stained with toluidine blue. (A) Lesions in media-only-injected controls were lightly stained reflecting the absence of myelin. (B) Higher magnification revealed lesions dominated by dark, myelin-filled macrophages and demyelinated axons (inset) with no remyelination. (C, E, G) Abundant remyelination was observed in lesions transplanted with embryonic, neonatal, and adult OECs. (D, F, H) Remyelination was widely distributed and densely packed. This remyelination has the characteristic “signet ring” morphology of peripheral myelin (D, F, H insets). Scale bars: 200 μm (A for C, D, F), 50 μm (B for D, F, H), and 2 μm (inset in B for insets in D, F, H).
Remyelination Is Most Extensive Following Transplantation of Embryonic OECs
Next, the question of which of the three donor ages of OECs led to the most extensive peripheral type myelination following transplantation was explored. Representative toluidine blue-stained semithin resin sections from all animals were examined by two researchers blinded to the group from which the section was obtained and ranked according to the extent of peripheral type remyelinated axons. The highest rank was given to the section(s) that exhibited the most extensive remyelination and lowest proportion of demyelinated axons (Fig. 4). After unblinding, control animals with media-only injections were ranked as having the least remyelination (indeed, none was detected). Remyelination following transplan tation of OECs from embryonic donors was ranked as significantly higher than that following transplantation with OECs from either neonatal or adult donors (p ≤ 0.05). There was no difference in the extent of remyelination by neonatal and adult OECs. Therefore, the maximum remyelination per quantity of cells transplanted was achieved by embryonic OEC transplant.

Enhanced remyelination by embryonic OECs. Comparison of extent of remyelination by medium-only controls, embryonic, neonatal, or adult OEC transplants. Representative toluidine blue-stained resin sections from each animal were coded to blind two observers to preparation type and ranked in order of completeness of remyelination. The highest ranking of 26 indicates the most complete remyelination. No remyelination was observed in media-only controls. Remyelination was observed in all donor ages and was significantly enhanced in embryonic transplants. ∗p ≤ 0.05; ns, not significant, Kruskal– Wallis test with Dunn's post hoc test.
Labeled OECs From Each Donor Age Remyelinate CNS Axons
Despite the lack of remyelination seen in control, nontransplanted spinal lesions in this experiment and when xenografted OECs are rejected, it nevertheless remains a possibility that the peripheral-type myelin observed within lesions following OEC transplantation could be mediated by host-derived cells encouraged into the lesion from outside the X-irradiated zone by the transplanted cells. The ultrastructural similarities between OEC and Schwann cell myelin mean that this possibility cannot be excluded by anatomical analysis. To determine whether the transplanted OECs had a direct or indirect role in the myelination seen following transplantation, prior to transplant, OEC cultures from each age were labeled using a GFP-expressing lentivirus to follow their fate in vivo.
Three weeks after transplantation, fluorescence microscopy revealed that GFP-positive cells were distributed throughout lesions (Fig. 5A). Immunopositive rings labeled with an antibody to the peripheral myelin-specific protein P0 were widely distributed throughout lesions in a similar pattern to identified peripheral-type myelin in resin sections. In transplants of OECs from all ages, peripheral myelin rings were frequently observed intimately associated with GFP-labeled transplanted OECs (Fig. 5B, C, D, insets). The subcellular location of P0 staining and proximity of a nucleus strongly suggests that the myelin sheaths were produced by the GFP-labeled cells, thus indicating that at least some of the peripheral-type myelin seen in resin sections was OEC derived.
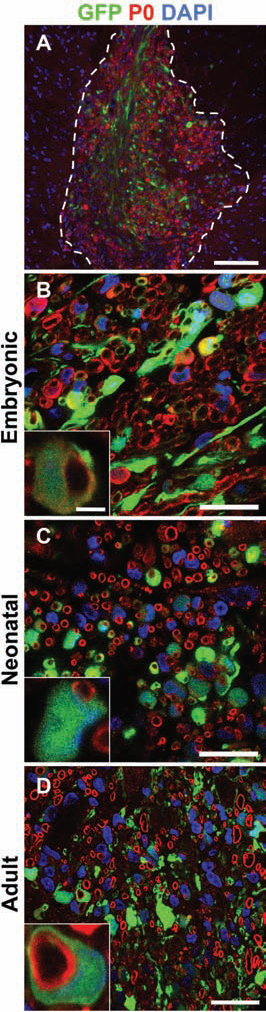
Green fluorescent protein (GFP)-labeled OECs form peripheral myelin sheaths. Transverse sections through lesions transplanted with GFP-labeled OECs. (A) GFP-labeled transplanted OECS and protein zero (P0)-positive peripheral myelin rings were distributed throughout the lesion (dashed line). (B–D) GFP-labeled OECs from embryonic, neonatal, and adult donors were frequently but totally associated with P0-positive myelin rings. GFP-positive cells containing Hoechst-stained nuclei were often observed ensheathing P0-positive myelin rings in a “signet ring” manner typical of peripheral myelinating cells (B–D insets). Intimately associated GFP and P0 myelin rings consisted 49.65 ± 0.65%, 52.01 ± 3.78%, and 53.58 ± 8.27% of total P0 myelin rings in embryonic-, neonatal-, and adult-transplanted lesions, respectively. Scale bars: 200 μm (A), 20 μm (B–D), and 2 μm (inset in B for C, D).
Labeled and Unlabeled OECs Are the Main Contributors to Remyelination
The question arose as to the origin of the P0+ myelin sheaths that were not associated with GFP+ cells. One possibility was that cells of endogenous host origin generated some of the peripheral myelin sheaths. An alternative explanation was that cells expressing GFP at the time of transplantation were no longer GFP+ at the end of the experiment 3 weeks after transplantation. Lentiviral vector infection with a multiplicity of infection (MOI) of 50 resulted in robust expression of GFP in 89–91% of OECs within 48 h of infection (Fig. 6A, B). In aliquots of transplanted OECs maintained in vitro for 3 weeks alongside the in vivo experiment, GFP expression was detectable, but was of variable intensity (Fig. 6C, D). Some cells displayed intense perinuclear staining (Fig. 6C, arrows), and the majority displayed at least weak cytoplasmic staining. Overall GFP expression had significantly decreased to around 69% on average (t test, p ≤ 0.001) (Fig. 6E). There was no significant difference in GFP expression between groups at transplant or after 3 weeks in culture (one-way ANOVA, p = 0.43) (Fig. 6E). To test whether GFP downregulation explained the presence of unlabeled P0+ myelin sheaths in lesions, the proportion of GFP+ to GFP- cells associated with P0+ myelin sheaths within the lesions was compared to the proportion of GFP+ and GFP- OECs kept in culture. In lesions examined 3 weeks posttransplant, approximately half of P0+ peripheral myelin sheaths were GFP+ (Figs. 5 and 6E). This was significantly less than the GFP expression in cells at transplant (t test, p ≤ 0.001) and after 3 weeks in vitro (t test, p ≤ 0.001) (Fig. 6E). This implies that while the majority of P0 myelin sheaths within the lesions were of transplant origin, a contribution of endogenous Schwann cells to remyelination cannot be ruled out.
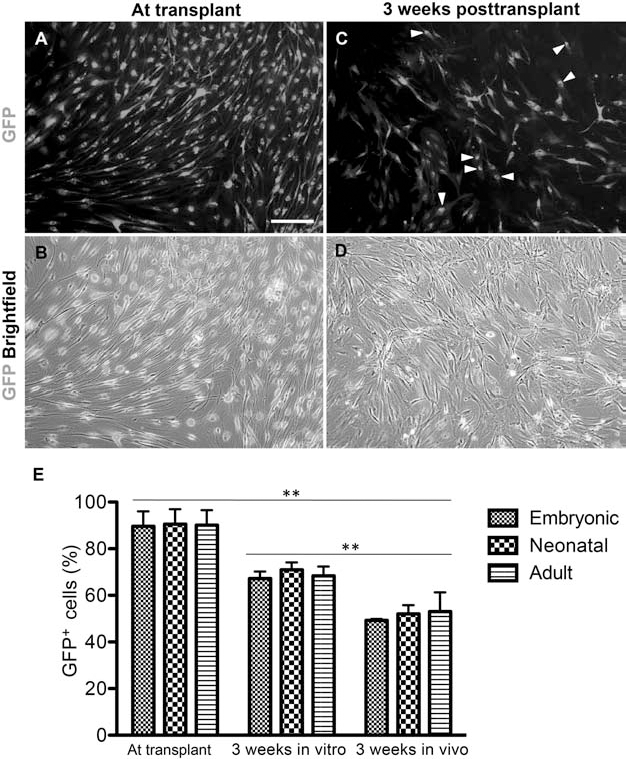
GFP lentiviral-labeled OECs in vitro and in vivo. (A, B) Strong nuclear and cellular GFP fluorescence was detected within 48 h after infection, at the time of transplant. (C, D) In aliquots of transplanted cells kept in culture for 3 weeks alongside the in vivo experiment, GFP fluorescence was weaker though still detected, with some cells showing strong perinuclear fluorescence (arrowheads). Scale bar: 200 μm. (E) Percentage of total GFP+ cells at time of transplant and after 3 weeks in vitro, and percentage of total P0+ myelin rings associated with GFP. ∗∗p ≤ 0.01.
Enhanced Embryonic OEC Remyelination Is Not Attributable to Enhanced Recruitment of Endogenous Schwann Cells
In lesions transplanted with GFP-labeled OECs, not all P0+ myelin sheaths were associated with GFP+ cells, indicating that endogenous Schwann cell remyelination may play a role in remyelination. The question arose as to whether the increased remyelination by embryonic versus neonatal and adult OECs was due to a greater propensity for the transplanted cells to engage directly in remyelination or indirectly by recruiting host-derived cells with peripheral myelinating potential. To address this, the proportion of GFP+/P0+ cells to P0+ myelin sheaths in each group was compared. GFP+/P0+ cells accounted for 49–54% of the total P0+ myelin sheaths, and this proportion was not significantly different between the groups (one-way ANOVA, p = 0.62). This indicated that the increased remyelination in the embryonic OEC transplanted group was mainly attributable to an increased remyelination propensity of the transplanted cells.
Discussion
Here we show that transplanted embryonic OECs can myelinate the demyelinated rat spinal cord. Under the same conditions, we find that neonatal and adult OECs are also fully capable of myelination. Remyelination is optimal when embryonic OEC transplants are used. This evidence is consistent with studies showing that OECs can myelinate in some in vivo studies (12,28) but not with other in vivo studies (6,23,32) or studies conducted in vitro (24).
A difficulty in comparing results of studies from different research groups is the differences in the experimental strategy employed. In this study, variation was minimized by employing an identical experimental strategy to investigate the effect of donor age on OEC myelination capacity. Olfactory bulb cultures from each donor age were immunopanned with an anti-p75NTR 192 IgG antibody, a method widely used to generate highly pure preparations of p75NTR-expressing OECs. Similarly, pure OEC cultures were required to accurately compare myelinating ability as different levels of cellular contaminants such as meningeal cells can modulate remyelination by OECs (21). Studies when purification did not occur invite speculation as to whether OECs are truly responsible for the conclusions drawn. Boyd and colleagues, using unpurified lacZ-labeled olfactory bulb cultures from embryonic rats, were unable to show any lacZ-labeled cells remyelinating axons following transplant into the contused spinal cord, while numerous nonlabeled cells formed peripheral type myelin (7). From this, they suggested that OECs do not myelinate and that contaminating or invading Schwann cells may be responsible for the peripheral myelin seen in other studies. The present study used the inbred, syngeneic Fischer-344 rat strain to avoid graft rejection. The study by Boyd and colleagues could not ensure this as it used the outbred, nongenetically identical Wistar rat with no immunosuppression leading to speculation of whether transplanted cells survived, although labeled cells (presumably of transplant origin) were identified. Therefore, rather than an inability of OECs to myelinate, differences in OEC preparation and most especially the microenvironmental signals to which they are subjected in the different experimental approaches both in vivo and in vitro are likely to account for the conflicting outcomes observed.
No remyelination was seen in control animals sham-transplanted with media only. This implies that the results of this study were not due to failure of the EB-X model causing endogenous remyelination by central or peripheral glia unaided by OEC transplant. However, it has been suggested that, rather than myelinating themselves, OEC transplant in the EB-X model enhances the invasion of endogenous Schwann cells that produce peripheral myelin (6). The results of this study show that embryonic GFP-labeled OECs are intimately associated with identified peripheral-type myelin in a manner highly suggestive of myelination (29). It seems likely therefore that the labeled peripheral myelinating embryonic cells were OECs (although we cannot entirely exclude the possibility of our preparations containing another undetected cell type capable of generating peripheral myelin sheaths). It is possible that transplanted OECs are tightly ensheathing endogenous myelinating Schwann cells as GFP signal and peripheral myelin staining rarely completely overlap. However, this cell arrangement is absent from ultrastructural analysis in any group and is more likely explained by the exclusion of GFP from compact myelin (29).
The data presented in this study confirm that OECs can myelinate. However, following transplant of GFP-labeled OECs, not all P0+ peripheral myelin sheaths were associated with GFP+ OECs. This may suggest that invasion of the lesion by endogenous Schwann cells contributes to some remyelination. Alternatively, GFP labeling may have failed or been downregulated in transplanted OECs responsible for GFP-negative remyelination. Ex vivo transduction inefficiency or instability leading to transgene downregulation is a well-known obstacle for long-term tracking of cell fate using viral vector-mediated gene transfer (11). Lentiviral transfection was chosen for its robust and sustained transgene expression (27); however, downregulation of transgene expression following lentiviral vector infection has been observed in neural cell types (11,20). A degree of reduced cytoplasmic expression was observed in lentiviral-transduced OECs kept in vitro, suggesting this may also be the case following transplantation. The proportion of GFP+-associated P0+ myelin sheaths in transverse sections of transplanted lesions was less than the proportion of GFP+ OECs kept alongside in vitro. This difference may suggest that endogenous Schwann cell invasion and myelination occurred. Alternatively, transverse sections of transplanted lesions may underestimate the proportion of GFP-labeled cells associated with P0+ myelin sheaths due to weak GFP signal in cytoplasmic regions further from the cell nucleus. Furthermore, inferences drawn from in vitro aliquots must be taken with caution as it does not take into account the effect exerted by the lesion environment. For example, GFP labeling may impact on cell survival within lesions or impair proliferation, reducing the proportion of myelinated axons associated with GFP+ cells. The lack of markers to distinguish OECs from Schwann cells means that it is not currently possible to determine the presence and extent of endogenous Schwann cell remyelination.
No differences in the proportion of GFP+ cells associated with P0+ peripheral myelin sheaths were observed between embryonic, neonatal, and adult OEC-transplanted lesions. This indicated that the increased remyelination in the embryonic OEC-transplanted group was mainly attributable to their increased remyelination propensity, rather than an increased ability to attract endogenous Schwann cells over neonatal or adult OECs. The mechanisms of enhanced remyelination are unclear. However, other myelinating glia show reduced myelinating capacity with ageing (30,34), although even in the CNS, myelination will eventually occur in aged animals albeit at a slower rate. No differences in remyelination capacity were observed between neonatal and adult OECs. This may indicate that remyelination capacity does not change or that differences were too subtle to be detected using this analysis. Recent evidence suggests that the latter may be more likely, as adult OECs exhibited slower myelination compared to juvenile OECs in vitro (1). In the current experiment, we cannot exclude the possibility that the differences in myelination between embryonic and neonatal/adult cells is one of rate rather than extent and that the older cells will eventually “catch up.”
Further work is required to determine whether the increased remyelinating capacity of embryonic OECs extends to other OEC-mediated regenerative mechanisms, such as axon outgrowth and sparing, improved angiogenesis and functional recovery (31). Furthermore, work is required to understand why embryonic OECs have a better remyelinating capacity than older cells. Embryonic cells may have increased migration ability, as embryonic-derived Schwann cell precursors can integrate better than mature Schwann cells following transplant into the CNS (35). Embryonic cells may also have a proliferation rate above that of neonatal or adult OECs, enabling faster repopulation of the lesion site with remyelinating cells. While this question could not be addressed with the single survival timepoint in this experiment, embryonic OECs required passaging in almost half the time as older cells, suggesting that proliferation rate may be a contributing factor to their enhanced remyelination capacity. Furthermore, adult rodent OECs undergo early senescence; however, adult non-human primate OECs are much more resistant to senescence (26). It remains to be determined whether the effect of age on remyelinating potential is similar for OECs from all species.
Our study has important implications for future study designs that will optimize the regenerative potential of OEC transplant. Speedy and complete remyelination is important to restore axonal function and protect demyelinated axons from irreversible atrophy (13). The potential for optimal regeneration by embryonic OECs must be carefully weighed against the ethical considerations of using human embryonic tissue and the requirement for immunosuppressive drugs for the remainder of life if a nonautologous donor source is used. However, recent identification of the developmental origins of OECs suggests that an autologous source of OECs with embryonic-like characteristics may be possible. Barraud and colleagues (4) show that OECs originate in the neural crest, like Schwann cells. This similarity underlies the often-reported difficulty in distinguishing cultured or transplanted OECs from Schwann cells—they have the same origins and antigenic profile yet behave in a different way determined by their developmental context. Neural crest stem cells can be sourced from hair follicles and can be converted into Schwann cells (15). If the cues that result in neural crest cells becoming OECs during development can be understood, adult neural crest stem cells may represent the ideal autologous source of embryonic-like OECs for transplant-mediated CNS repair. In conclusion, this study supports the use of OEC transplants for myelin-mediated repair and suggests that embryonic OECs are optimal to achieve remyelination.
Footnotes
Acknowledgments
This work was supported by grants from the UK MS Society and ARSEP (RJMF) and Western Australian Neurotrauma Program (GWP). DJCC was funded by a Gates Cambridge Scholarship. CEH was funded by an Athelston and Amy Saw Medical Research Fellowship. We would like to thank Mike Peacock for technical assistance.