
Abstract
Multiple sclerosis (MS) is a severe debilitating disorder characterized by progressive demyelination and axonal damage of the central nervous system (CNS). Current therapies for MS inhibit the immune response and demonstrate reasonable benefits if applied during the early phase of relapsing–remitting MS (RRMS) while there are no treatments for patients that progress neither to the chronic phase nor for the primary progressive form of the disease. In this manuscript, we have studied the therapeutic efficacy of a cell and gene therapy strategy for the treatment of a mouse model of chronic MS [myelin oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis (EAE)]. We used allogenic mesenchymal stem cells (MSCs) as a therapeutic tool and also as vehicle to deliver fully processed 3.3-kDa vasoactive intestinal peptide (VIP) to the peripheral immune organs and to the inflamed CNS. Intraperitoneal administrations of MSCs expressing VIP stopped progression and reduced symptoms when administered at peak of disease. The improvement in clinical score correlated with diminished peripheral T-cell responses against MOG as well as lower inflammation, lower demyelination, and higher neuronal integrity in the CNS. Interestingly, neither lentiviral vectors expressing VIP nor unmodified MSCs were therapeutic when administer at the peak of disease. The increased therapeutic effect of MSCs expressing VIP over unmodified MSCs requires the immunoregulatory and neuroprotective roles of both VIP and MSCs and the ability of the MSCs to migrate to peripheral lymph organs and the inflamed CNS.
Keywords
Introduction
Multiple sclerosis (MS) is a severe debilitating disorder characterized by progressive demyelination and axonal damage of the central nervous system (CNS). Although the exact causes that trigger disease development are not known, it is generally accepted that MS starts when autoreactive T-cells infiltrate the CNS and mount a demyelinating immune attack (28). MS usually begins as a relapsing–remitting (RRMS) disease, which in about 65% of the cases develops into secondary-progressive MS (SPMS). RRMS is characterized by periods of acute disability followed by periods of functional recovery. In this form of the disease, the resolution of inflammation is followed by remyelination of damaged axons and partial or complete recovery of the symptoms. However, SPMS is associated with chronic microglia activation and a steady disease progression due to heavy axonal loss and neurodegeneration. In general, the currently approved MS therapies [e.g., interferon-β (IFN-β), glatiramer acetate, and mitoxantrone], inhibit the immune response and demonstrate thus greater benefits on RRMS compared to SPMS, postponing the secondary progressive phase. Approved therapies of MS are also associated with side effects including depression, multifocal leukoencephalopathy, and hypersensitivity reactions warranting the development of safer and more effective therapies, especially for SPMS.
Mesenchymal stem cells (MSCs) have emerged as a promising therapy of MS because of their ability to migrate into sites of inflammation, suppress the immune response, and protect neuronal cell death through the release of neuroprotective factors or possibly through transdifferentiation of MSCs into neurons or oligondendrocytes (4,21,46,56,65). Bone marrow-derived MSCs (BM-MSCs) have traditionally been used in most preclinical and clinical trials, although other potential sources are umbilical cord, endometrial polyps, menses blood, peripheral blood, and adipose tissue (14). Adipose tissue-derived MSCs are emerging as an attractive replacement because of their promising therapeutic effects and readily available, noninvasive source in using lipoaspirates (27). Adipose tissue-derived MSCs resemble their bone marrow-derived counterpart, but some differences in gene expression profile, phenotype, and proliferative capacity exist. Importantly, adipose tissue-derived MSCs possess an equal or more potent immunosuppressive capacity compared to BM-MSCs (52) and inhibit pathologies in several animal models of autoimmunity and inflammation (24,27).
Experimental autoimmune encephalomyelitis (EAE) is a clinically relevant model of MS, which has given rise to several approved MS therapies, including IFN-β (60). EAE and MS share both clinical and pathological features, including infiltration of activated autoreactive T-cells into the central nervous system (CNS), axonal injury, and cortical demyelination as well as multiple CNS auto antigens [myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), proteolipid protein (PLP)]. However, there are also important differences between EAE and MS. For instance, in EAE most of the lesions have predominantly cluster of differentiation 4 (CD4+) T-cells, while in MS CD8+ T-cells and macrophages are the most abundant (31). Of particular relevance for our study is the MOG-induced EAE in C57Bl/6 mice. The pathology induced in this model is self-limited, monophasic, and chronic in which there exist a correlation between lesion development and disease severity. In addition, glial fibrillary acidic protein (GFAP) is upregulated in chronic EAE and in secondary progressive MS, but not in relapsing–remitting MS (37).
Systemic administration of MSCs has shown to ameliorate EAE by inhibiting the expansion and cytokine production by autoreactive T-helper (Th)1 and Th17 cells resulting in a decrease in CNS demyelination (8). However, whereas MSC administration readily ameliorates disease symptoms in murine EAE, human trials for the treatment of MS have given less impressive results. The number of MSCs injected is most likely an important factor contributing to the discrepancy. In animal studies, the normal dose is 40–80 × 1 0 6 cells/kg, whereas in human trials, the cell number has averaged 2 × 1 0 6 cells/kg due to limitations in the expansion potential of MSCs in vitro. Increasing the potency of MSCs through forced expression of therapeutic genes could be a step in the right direction toward a successful MSC-based therapy of MS.
Several groups have attempted to increase therapeutic potential of MSCs by expressing different genes (43–45). The aim is to enhance the homing capacity and increase the immunomodulatory and neuroregenerative potential of MSCs by delivering new therapeutic agents to areas of inflammation. In this strategy, MSCs have two roles: [1] to deliver the therapeutic transgene to peripheral immune tissues and to the damaged CNS and [2] to make use of its natural immunomodulatory and neuroprotective capabilities to protect the damaged CNS. For instance, MSCs/interleukin-10 (IL-10) prevented lung ischemia-reperfusion injury (41), MSCs/neurotrophin-3, MSCs/bone morphogenetic protein (BMP)-9, and MSCs/BMP-2 all improved spinal fusion (15,45,63), MSCs/angiopoietin −1 + vascular endothelial growth factor (VEGF) and MSC/placental growth factor (PlGF) improved cerebral ischemia (38), MSC/chemokine (C-X-C motif) receptor 4 (CXCR4) improved postinfarction myocardial repair (6), MSC/transforming growth factor-β1 (TGF-β1) and MSC/BMP-2 improved cartilage repair (48,49), and MSC/brain-derived neurotrophic factor (BDNF) promoted functional recovery and reduce infarct size in a rat model of middle cerebral artery occlusion (36).
Vasoactive intestinal peptide (VIP) is a neuropeptide of 28 amino acids with a broad spectrum of biological activities including immunoregulation (dampening T-cell responses and lowering inflammation) and neuroprotection (blocking microglial activation and induction of neuroprotective factors) (29). We and others have demonstrated important therapeutic effects of systemic delivery of synthetic VIP in several animal models of autoimmune diseases (10). In addition, a recent phase I/II clinical trial in patients with sarcoidosis demonstrated that inhalation of VIP decreased the levels of inflammatory markers in lung and increased the number of suppressive regulatory T-cells (Tregs) (51). However, multiple doses of high concentration of the peptide are required to obtain the beneficial effects. In addition, the instability of the peptide makes this strategy difficult to perform and highly variable, especially when VIP is systemically administered. In order to improve delivery of VIP into target organs, we have previously developed a lentiviral vector expressing VIP cDNA (LentiVIP) (13). A single injection of LentiVIP in a mouse model of severe rheumatoid arthritis (RA) provided a highly effective treatment with complete regressions in established disease associated with reduction of the autoimmune and inflammatory responses. In another study, we developed tolerogenic dendritic cells (DCs) by transducing DCs with LentiVIP (DCs/LentiVIP) during differentiation from bone marrow cells (57). A single administration of DCs/LentiVIP proved therapeutic when administered before onset of EAE by reducing IL-1β, tumor necrosis factor (TNF)-α, and IL-6 and increasing IL-10. However, in spite of their potential, the difficulties to scale up DC/LentiVIP production could be a serious limitation for future clinical applications of this strategy. In addition, while allogeneic cells are a very interesting alternative when considering cell therapies as potential medicaments, the use of allogeneic DCs/LentiVIP is not an alternative due to their potential side effects. We have therefore looked for alternative therapeutic strategies aiming to treat MS.
In the present manuscript, we have studied the therapeutic efficiency of adipose tissue-derived MSCs expressing VIP (MSCs/LentiVIP) for the treatment of a mouse model of chronic MS (MOG-induced EAE in C57Bl/6 mice) and compared it with the effects of direct injection of LentiVIP particles and unmodified MSCs. We obtained routinely over 108 MSCs/LentiVIP secreting VIP. We showed that direct injection of LentiVIP particles did not result in LentiVIP expression in CNS and was not effective for the treatment of chronic EAE. In contrast, injection of allogeneic MSCs/LentiVIP (but not the unmodified MSCs) into mice with severe established EAE ameliorated disease symptoms. The therapeutic efficacy of MSC/LentiVIP treatment correlated with a stronger suppression of the autoimmune T-cell response and less CNS inflammation/damage compared to treatment with MSCs/LentiGFP or with unmodified MSCs.
Materials and Methods
Animals
Male Balb/c (6–10 weeks) and female C57Bl/6 (6–8 weeks) (Charles River, Barcelona, Spain) were used to initiate cultures of adipose tissue-derived MSCs and for the induction of EAE, respectively. All experiments were performed according to the Institutional Guidelines for the Care and Use of Laboratory Animals in Research and the approval of the local committee in the Consejo Superior de Investigaciones Cientificas.
Plasmids and Lentiviral Constructs
The HIV packaging (pCMVΔR8.91) and VSV-G (pMD.G) plasmids were kindly provided by D. Trono (University of Geneva, Geneva, Switzerland). The packaging plasmid pCMVΔR8.91 encodes group-specific antigen (gag), polymerases (pol), transactivating regulatory protein (tat), and regulator of virion expression (rev) genes. The pMD.G plasmid encodes the vesicular stomatitis virus G (VSVg) glycoprotein. The LentiGFP (green fluorescent protein; CEWP) and LentiVIP lentiviral vectors express eGFP and VIP, respectively, through the strong cytomegalovirus containing tet operator (CMVTetO) promoter. They were constructed as previously described (13).
Isolation and Culture of MSCs
MSCs were isolated from abdominal fat from male Balb/c mice. The fat was aseptically removed and washed twice in Hank's balanced salt solution (HBSS; GIBCO, Invitrogen, Carlsbad, CA, USA). The fat tissue was weighed, cut into small pieces (≤2 mm3), and resuspended in 2.5 ml of HBSS containing 2 mg/ml collagenase type 1 (Sigma Aldrich, St. Louis, MO, USA) per gram fat tissue and incubated for 30 min at 37°C, swirling the tube every 5 min. The digest was washed twice with 10 ml HBSS and filtered each time through a 100-μm cell strainer [Becton Dickinson (BD), Franklin Lakes, NJ, USA]. The cells were washed with HBSS again and filtered through a 40-μm cell strainer. Finally, cells were resuspended in complete MesenCult (Stem Cell, Grenoble, France) medium containing 100 units/ml penicillin/streptomycin (Gibco, Invitrogen) and 20% mouse mesenchymal supplements (Stem Cell) and plated at a density of 15,000–30,000 cells/cm2 and cultured at 5% O2. Nonadherent cells were removed after 24 h in culture. Subsequent passages were plated at 5,000–10,000 cells/cm2 in complete MesenCult media. MSCs/LentiVIP and MSCs/LentiGFP were generated by transduction with LentiVIP and LentiGFP vectors, respectively.
Immunophenotypic Analysis and Foxp3 Staining
MSCs, MSCs/LentiVIP, and MSCs/LentiGFP were stained with allophycocyanin (APC)-labeled antibodies against CD45, CD44, stem cell antigen 1 (Sca1), major histocompatibility complex II (MHCII), and CD29 (eBioscience, San Diego, CA, USA). For forkhead box p3 (Foxp3) staining, cells from draining lymph nodes (DLNs) and spleens were isolated from mice 5 days posttreatment and incubated with 7-amino-actinomycin D (h7AAD; Sigma-Aldrich) and 24G2 (eBioscience) and stained with the FoxP3 staining kit (eBioscence) according to the manufacturer's instructions. Cells were acquired and analyzed on a BD FACS Canto II using the fluorescence activated cell sorting (FACS) Diva software (BD Biosciences, San Jose, CA, USA).
Vector Production and Titration
Lentiviral vectors were produced by cotransfection of 293T kidney cells (human embryonic kidney cells, CRL-11268, ATCC) with three plasmids: (1) Vector plasmid (CEWP or LentiVIP), (2) Packaging plasmid pCMVΔR8.91, and (3) Envelope plasmid pMD.G using Lipofectamine 2000 (Invitrogen) as previously described (58). Viral supernatants were collected and filtered through a 0.45-μm filter and immediately frozen at −80°C or used to transduce MSCs. Vector particles were concentrated by ultrafiltration at 2,000 × g and 4°C, using 100-kDa centrifugal filter devices as previously described (18) (Amicon Ultra-15, Millipore, Billerica, MA, USA). Vector titration was performed in 293T cells. CEWP vector titration was determined by the percentage of eGFP-positive cells by FACS analysis 7 days posttransduction. For titration of LentiVIP vectors, transduced cells were lysed and DNA was extracted after 7–10 days. Vector copy number per cell was determined using Q-PCR. Q-PCR was performed with MX3005Pro sequence detection system (Stratagene, Santa Clara, CA, USA). Primer pairs: VIP: FW 5′-CCA CGC TGT TTT GAC CTC CAT, RV 5′-GGG CCT TAT TTC TGG TGT CCA T.
Analysis of Secreted VIP
MSCs/LentiVIP, MSCs/LentiGFP, and MSCs were seeded in six-well plates or 25 cm2 flasks (20,000 cells/cm2). Supernatant were collected after 48 h and frozen at −80°C in 2 ml aliquots and cell lysed with 1% Nonidet P40 lysis buffer containing protease inhibitor cocktail (Sigma).
To detect the processed form of VIP, the supernatant from a 25 cm2 flask was concentrated by lyophilisation and fractionated by high-performance gel filtration in AKTA fast protein liquid chromatography (FPLC) system using Superdex 75 10/300 GL column (GE Healthcare Biosciences, Uppsala, Sweden). This technique allows the separation of proteins and peptides based on its molecular weight. We collected 36 fractions of 1 ml each. As controls, we collected the same fractions from a solution containing 2 ng/ml of VIP (Calbiochem, Laufelfigen, Switzerland). The different fractions were analyzed for VIP reactivity using an ELISA Kit (Phoenix Pharmaceuticals, Cat No. EK-064-16, Karlsruhe, Germany). Positive fractions were precipitated with trichloroacetic acid (TCA; Sigma) as previously described (42). Briefly, we added 200 μl of TCA to 800 ml of each fraction, incubated 30 min on ice and centrifuged at 13,200 rpm at 4°C. The pellet is washed with 1 ml of acetone for 1 min at RT and centrifuged for 20 min at 13,200 rpm. The supernatant was removed, and the pellet was resuspended in 50 μl of distilled water. Finally, we added 10 μl of SBR6X buffer [12% SDS (w/v); 30% β-mercaptoethanol; 60% glycerol; Tris- HCl 0.4 M, pH 6.8; bromophenol blue 0.03% (w/v); all Sigma], boiled at 95°C for 5 min and left on ice.
For Western blot analysis, the different samples (cell lysates, concentrated supernatants, and precipitated fractions) were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE; 15% polyacrylamide under reducing conditions) and electrotransferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% nonfat milk (Sigma) and probed for 1 h at room temperature with anti-VIP polyclonal antibody at 1:200 dilution (anti-VIP (H-16); Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by 1 h of incubation with donkey anti-goat antibody conjugated with the infrared dye 800 CW (1:10,000 dilution; LI-COR Biosciences, Cambridge, UK). Protein was detected using Odyssey Image scanner system (LI-COR).
EAE Induction and Treatment
C57Bl/6 mice were injected subcutaneously with 100 μg of MOG35–55 (Genescript, Hong Kong, China) emulsified in complete Freund's adjuvant (CFA) (Difco, Detroit, MI, USA) containing 0.5 mg of M. tuberculosis (Difco). Mice were injected intraperitoneally (IP) with 100 ng of pertussis toxin (Sigma-Aldrich), at the same day of immunization and 48 h later. Lentiviral treatment consisted of a single IP administration of 2 × 1 0 8 LentiVIP transduction units (tus) per mouse. As control, mice were injected with PBS or LentiGFP (2 × 108 tus/mouse). MSC treatment consisted in a single IP administration of 106 MSCs, MSCs/LentiGFP, or MSCs/LentiVIP per mouse when mice showed established disease (clinical score = 2.5–3.5). Clinical scores used are as follows: grade 0, no clinical signs; grade 1, complete loss of tail tonicity; grade 2, flaccid tail and abnormal gait; grade 3, hind leg paralysis; grade 4, hind leg paralysis with hind body paresis; grade 5, hind and fore leg paralysis; grade 6, death. Mice that were in between the clear-cut gradations of clinical signs were scored intermediate in increments of 0.5.
Proliferation Assays
To study the effect of MSCs on T-cell proliferation, MSCs were irradiated at 42 Gy and plated in triplicates in 96-well plates at 40,000, 20,000, 10,000, 5,000, and 0 cells/well. Splenocytes were isolated (erythrocytes were lysed) and added to the wells at 200,000 cells/well and stimulated with concanavalin A (ConA; 2.5 μg/ml) (Sigma). For the in vitro analysis of the MOG35–55-specific recall responses, cell suspensions were obtained from DLNs of mice 7 days after injection of MSCs. The cells were plated at 4 × 1 0 5 cells/well in triplicates and stimulated with MOG35–55 (50 μg/ml) and anti-CD3 (1 μg/ml) (eBioscience). In both cases, after 3 days, cells were pulsed with 0.5 μCi/well [3H]thymidine (Perkin Elmer, Waltham, MA, USA) for 6 h and harvested onto glass fiber filters using an LKB 96 well harvester (Wallac Oy, Turku, Finland). Uptake of [3H]thymidine was measured on a 1450 microbeta Trilux scintillation counter (Wallac).
Vector and Cell Distribution
The distribution of LentiVIP and MSCs/LentiVIP was studied by standard PCR. Primer pair: VIP: FW 5′-CAC CAC CTG TCA GCT CCT TT-3′; RV 5′-ACA ACA CCA CGG AATTGT CA-3′. PCR was performed by using Mastercicler EpgradientS (Eppendorf, Hamburg, Germany) and analyzed on agarose gels [1 g agarose in 100 ml Tris base, acetic acid, and EDTA (TAE) Sigma].
RNA Extraction and Quantification of Transcripts
The RNA from mouse tissues was obtained using trizol reagent (Invitrogen). RNA samples were reverse-transcribed into cDNA using Superscript first-strand (Invitrogen). Q-PCR was performed by using the QuantiTect SYBRGreen PCR kit (Qiagen, Madrid, Spain) in a Stratagene MX3005P system (Madrid, Spain). Primer pairs: IL-17: FW 5′-TTT AAC TCC CTT GGC GCA AAA-3′, RV 5′-CTT TCC CTC CGC ATT GAC AC-3′; inducible nitric oxide (iNOS): FW 5′-GTT CTC AGC CCA ACA ATA CAA GA-3′, RV 5′-GTG GAC GGG TCG ATG TC AC-3′; TNF-α: FW 5′-GGC AGG TCT ACT TTG GAG TCA TTG C-3′, RV 5′-ACA TTC GAG GCT CCA GTG AAT TCG G-3′; IL-10: FW 5′-CTG GAC AAC ATA CTG CTA ACC G-3′, RV 5′-GGG CAT CAC TTC TAC CAG GTA A-3′; IL-6: FW 5′-TAG TCC TTC CTA CCC CAA TTT CC-3′, RV 5′-TTG GTC CTT AGC CAC TCC TTC-3′; FoxP3: FW 5′-CCC ATC CCC AGG AGT CTT G-3′, RV 5′-ACC ATG ACT AGG GGC ACT GTA-3′; activity-dependent neuroprotective protein (ADNP): FW 5′-AGAAAAGCCCGGAAAACTGT-3′, RV 5′-AAGCAC TGC AGC AAA AAG GT-3′; BDNF: FW 5′-CCC TCC CCC TTT TAA CTG AA-3′, RV 5′-GCC TTC ATG CAA CCG AAG TA-3′.
Immunohistochemistry and Staining Quantification
Mice were killed 7 or 50 days after treatment, and brain and spinal cord were removed and immersed in OCT and frozen in chilled 2-methylbutane (Sigma) and stored at −80°C. Sections were cut at 4 μm and incubated with antibodies against GFAP (Dako, Glostrup, Denmark), CD11b (Millipore), β-amyloid (Sigma-Aldrich), β-tubulin (Millipore), MBP (Millipore). Sections were then stained with a goat anti-mouse phycoerythrin (PE)-conjugated IgG (Millipore) and DAPI (Vectashield, Vector, Peterborough, UK). Histological examination was done using a Nikon Eclipse fluorescence microscope (Melville, NY, USA). An average of 10 different images per section was captured using a Nikon digital sight controller with the NIS element BR 3.10 program (Nikon). The acquisition conditions were always set up to background levels related to staining samples with isotype controls. Each image was then analyzed to measure number of spots/cells. For each antibody, the appropriate isotype control was used to set up the object count to zero. Sections from healthy and EAE mice were used to set up the conditions to measure the numbers of the desire spots. For that, both the area (px2) and circularity parameters of the spots were restricted with the NIS element program to focus the quantification to the desired spots. Once the settings were established, all images (taken from tissue sections from the different treatments) were processed equally to determine number of spots.
Statistical Analysis
For statistical evaluation, clinical EAE score results are represented as median and analyzed using the non-parametric Kruskal–Wallis test followed by the Dunn's posttest (with a significance level of p ≤ 0.05). For the analysis of gene expression, proliferation and histology data results are expressed as mean (±SD) and analyzed using the Student's t test or ANOVA, followed by the Tukey–Kramer posttest (with a significance level of p ≤ 0.05). Results were considered significantly different if p ≤ 0.05.
Results
Direct Injection of LentiVIP Particles Into Mice with Established Chronic EAE Lack Therapeutic Effect
Repetitive administration of exogenous VIP has been shown to inhibit disease in both relapsing–remitting and chronic models of EAE in the mouse (26). However, VIP is rapidly subjected to proteolytic degradation in vivo substantially lowering potency and clinical applicability of exogenous peptide administration (22). In order to improve therapeutic efficiency of VIP for the treatment of autoimmune diseases, we have previously developed a lentiviral vector driving the expression of preproVIP cDNA (LentiVIP) (13) (Fig. S1; http://www.genyo.es/resources/figures.zip). A single injection of LentiVIP particles enhanced the therapeutic benefits achieved by VIP peptide in a collagen-induced arthritis (CIA) mice model of RA (13). We hypothesized that a similar strategy could also be applied to improve therapeutic strategies for MS. We induced chronic EAE in C57Bl/6 mice using the MOG35–55 peptide, and the animals were intraperitoneally inoculated with an average of 108 tus (transduction units) of LentiVIP particles at the peak of disease (mice with an EAE score of 3–3.5; see scoring in Materials and Methods). Contrary to what we found in the CIA model, we could not observe any significant therapeutic effect when the LentiVIP particles were administered at the peak of disease (Fig. 1a, b). We detected vector genomes and vector-derived VIP expression in spleen, draining lymph nodes (DLNs), and liver (Table 1), but we were unable to detect the vector in neither the brain nor the spinal cord.

Direct injection of lentiviral particles expressing VIP has no effect on established EAE. (a) Graph showing experimental autoimmune encephalomyelitis (EAE) progression after injection of 108 lentiviral-vasoactive intestinal peptide (LentiVIP) particles (■) at the peak of the disease (3–3.5). Untreated mice (□) and mice treated with LentiGFP (lentiviral-green fluorescent protein) (Δ) showed identical EAE progression, indicating the failure of LentiVIP administration for EAE therapy at this stage of disease. Data are shown as mean ± SD of 5 (untreated and LentiGFP) and 9 (LentiVIP) animals per group. (b) Scatter plot showing the score of individual mice and the median for each group 31 days posttreatment. No significant differences were found between untreated and LentiVIP-treated animals as determined by the Kruskal–Wallis test.
LentiVIP Transcripts Are Detected at the CNS of EAE Mice Only When MSCs/LentiVIP Are Inoculated

Presence (+) or absence (–) of vasoactive intestinal peptide (VIP) mRNA sequences derived from LentiVIP was determined using standard PCR techniques (see Materials and Methods). Mice were inoculated with 108 LentiVIP particles or 106 mesenchymal stem cells (MSCs)/LentiVIP and killed 7 days later for analysis. ND, not determined; EAE, experimental autoimmune encephalomyelitis; DLN, draining lymph nodes.
MSCs/LentiVIP Secrete Fully Processed VIP While Maintaining the Main Properties of MSCs
In order to achieve an improved MS therapy, we designed a combined gene cell therapy strategy in which we wanted to combine the anti-inflammatory and neuroprotective properties of VIP with two main characteristics of MSCs: (1) the migratory potential of MSCs toward inflamed tissues (acting as a vehicle to deliver VIP to the damaged CNS) and (2) the immunoregulatory and neuroprotective properties. Nontransduced MSCs did not express nor secrete VIP, neither under resting conditions nor after stimulation with lipopolysaccharide (LPS) or inflammatory cytokines (Fig. S2, data not shown; http://www.genyo.es/resources/figures.zip). Transduction of MSCs with LentiVIP (MSCs/LentiVIP) at multiplicity of infection (MOI) = 3 resulted in the expression and secretion of 1–3 ng/ml of VIP as detected by ELISA (Fig. S2; http://www.es/resources/figures.zip). VIP mRNA is first translated into a 170 aa (19.2 kDa) precursor peptide (preproVIP), which is then convertases process by proteolysis, generating the fully processed form of VIP (28 aa and 3.3 kDa) (16). However, we could not detect secreted VIP in the supernatant of these MSCs/LentiVIP using Western blot due to the poor sensitivity of detection (data not shown). We therefore transduced MSCs with a high dose of LentiVIP (MOI = 100) to express high levels of VIP (MSCs/LentiVIPhd). Western blot of cell lysates and supernatant of MSCs/LentiVIPhd demonstrated that the 19.2-kDa preproVIP was the main VIP secreted by these cells (Fig. 2a). Other polypeptides with lower molecular weight were also visible. However, we could not detect the 3.3-kDa VIP in either MSC/LentiVIPhd supernatant or in cell lysates. In order to determine whether MSCs/LentiVIPhd secreted the fully processed form of VIP, we separated the supernatant in different fractions (based on protein size) using high-performance gel filtration (see Materials and Methods for details) and analyzed VIP content in the collected fractions by ELISA (Fig. 2b, graph). Contrary to the data obtained by Western blot of total supernatant (Fig. 2a), these experiments seemed to indicate that the majority of the secreted VIP was fully processed since the fraction with the highest concentration of VIP (114 ng/ml) contained only the fully processed 3.3-kDa VIP (Fig. 2b, bottom; F15). The discrepancies between the data obtained with the ELISA (Fig. 2b) and Western blot of total supernatant (Fig. 2a) can be explained if the ELISA Kit from Phoenix Pharmaceuticals is highly specific for the processed form of VIP. Indeed, we support this hypothesis since MSCs/LentiVIP secreted mostly preproVIP (Fig. 2a), and still the ELISA kit detected almost exclusively the 3.3 kDa VIP peptide (Fig. 2b). All unprocessed VIP, including the fractions containing preproVIP (F7 and F9), are only poorly recognized (Fig. 2b, graph of Fractions F7 and F9). By contrast, the Western blot using the H16 polyclonal antibody was more efficient at detecting preproVIP than the 3.3-kDa VIP. In summary, we have concluded that MSCs/LentiVIP secrete fully processed VIP although in much lower quantities compared to unprocessed preproVIP.

LentiVIP-transduced mesenchymal stem cells (MSCs/LentiVIP) stably secrete fully processed VIP and maintain the T-cell inhibitory potential of non-transduced MSCs. (a) MSCs/LentiVIP secreted several forms of unprocessed VIP. MSCs were transduced with a high dose of LentiVIP [multiplicity of infection (MOI) = 100; MSC/LentiVIPhd] in order to detect all forms of VIP. Cell lysates (CL) and supernatants (Sup) from MSCs/LentiVIPhd were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis-(SDS-PAGE) under reducing conditions and transfer to nitrocellulose membranes. The presence of VIP was analyzed using a polyclonal antibody recognizing VIP and a secondary donkey anti-goat antibody conjugated with the infrared dye 800 CW. 10ng of VIP peptide (VIP) was loaded as control. Only unprocessed (19.2 kDa) and partially processed forms of VIP (10–12 kDa) were detected using this technique. (b) MSCs/LentiVIP secrete the fully processed 3.3-kDa VIP peptide. Supernatant of MSCs/LentiVIPhd were concentrated by lyophilization (5x) and separated in different fractions by high-performance gel filtration (see Materials and Methods). VIP concentration of each fraction was determined by ELISA kit. The fraction with higher VIP content (Fraction 15) and representative positive fractions corresponding to higher molecular weight (Fractions 7, 9, and 11) were precipitated with trichloroacetic acid (TCA), resuspended and analyzed by Western blot (bottom). Of note, the fraction with higher concentration of VIP (114 ng/ml, as determined by ELISA) contained only the fully processed form of VIP. (c) Analysis of VIP transgene expression over time in MSC/LentiVIP lines. MSC lines transduced with LentiVIP at high (left) or low MOI (right) were analyzed with real-time quantitative (Q)-PCR at passages 5, 8, and 15 (5p, 8p, and 15p, respectively). Data represent arbitrary units relative to untransduced MSCs. (d) Graph showing stability of synthetic VIP peptide (left) versus MSC/LentiVIP-secreted VIP (right). VIP (added to the conditioned medium of non transduced cells) (left) and conditioned medium from MSCs/LentiVIP (right) were harvested and incubated at 37°C for up to 48 h. The concentration of VIP was measured at different time points by a VIP-specific ELISA. Data shown are mean ± SD. (e) MSCs/LentiVIP inhibit T-cell proliferation at a similar level to nontransduced MSCs. Different numbers (0–40,000 cells) of irradiated MSCs (♦), MSCs/LentiVIP (■), and MSCs/LentiGFP (▲) were added to a fixed amount (200,000 cells) of concanavalin A (2.5 μg/ml) stimulated splenocytes. Proliferation was measured by thymidine incorporation (see Materials and Methods). NS, non stimulated splenocytes (Δ).
Expression of VIP was maintained in MSCs/LentiVIP over time in culture (Fig. 2c) and, interestingly, the MSC-secreted VIP was more stable than the synthetic VIP peptide at 37°C when both were incubated in the same media (Fig. 2d, left vs. right graph). Since the ELISA was mainly detecting 3.3-kDa VIP, these data indicate that the processed VIP secreted by the MSCs/LentiVIP is more stable than synthetic VIP. This could be explained by posttranslational modification such as carboxyamidation and/or the addition of glycines (16).
Independent of the expression levels, the MSC/LentiVIP lines maintained their main phenotypic characteristics in terms of morphology, differentiation potential, and expression of membrane markers. All MSC populations expressed similar levels of Sca-1, CD29, and CD44 while lacking expression of CD45 and MHC class II (Fig. S3; http://www.genyo.es/resources/figures.zip). MSCs inhibit T-cell proliferation induced by anti-CD3/CD28 antibodies, ConA, and mixed leukocyte reactions (MLRs) (64). VIP has also been shown to inhibit the proliferation of and cytokine production by murine T-cells (47) and to prevent the maturation of dendritic cells (12). In order to assess a possible contribution of VIP to the MSC-mediated T-cell suppression, we activated splenocytes with ConA in the presence or absence of increasing numbers of non-transduced MSCs, MSCs/LentiGFP, or MSCs/LentiVIP. All MSC populations inhibited splenocyte proliferation to the same extent in a dose-dependent manner (Fig. 2e). We did observe a small but consistent increase in suppressive activity in MSCs expressing VIP, but the difference did not reach statistical significance.
A Single Dose of MSCs/LentiVIP at the Peak of Disease Improves Survival and Reduces EAE Severity
Next, we wanted to assess the potential of VIP-expressing MSCs on established disease in which both immunomodulation and neuroprotection would be beneficial. Most studies on the therapeutic effect of MSCs on EAE have administered the MSCs before or early after disease onset (53). However, once the disease is approaching its peak, many therapies lose their beneficial effects as have been seen in both mouse studies and with human MS drugs (19,59). We therefore explored the therapeutic potential of allogeneic MSCs/LentiVIP in the MOG35–55-induced EAE model of chronic MS at the peak of the disease (average score = 3.3; complete paralysis of back legs and partial paralysis of front legs). We selected cell lines secreting about 2 ng/ml of VIP and expanded them for four to five passages. EAE mice were injected intraperitoneally with 106 MSCs/LentiVIP, MSCs/LentiGFP, or non-transduced MSCs. Only MSCs/LentiVIP were able to significantly reduce EAE severity (Fig. 3a, b and Table 2; see also Fig. S4; http://www.genyo.es/resources/figures.zip), while most of the mice treated with MSCs alone or MSCs/LentiGFP progressed from moderate to severe EAE. This was corroborated when the cumulative disease index (CDI) was determined: MSCs/LentiVIP decreased CDI to 131 ± 32 (p = 0.001) in comparison to untreated (245 ± 60), MSC-treated (226 ± 65), and MSC/LentiGFP-treated mice (227 ± 74) (Table 2). The MSC/LentiVIP-treated mice recovered from complete hind leg paralysis toward a moderate hind leg paresis, whereas the remaining groups developed a more aggressive disease (Fig. 3a, b and Table 2). Of note, none of the MSC/LentiVIP-treated mice were required to be killed due to deterioration by the disease, while 50%, 60%, and 70% of the mice were treated with MSCs, MSCs/LentiGFP, and PBS, respectively, were killed or died from EAE severity (Table 2). MSC-derived VIP could be detected in the spleen, liver, kidney, and DLNs (Table 1). Importantly, in contrast to the LentiVIP particles, MSC-derived VIP could be detected in both brain and spinal cord (sc) (Table 1 and Fig. S5; http://www.genyo.es/resources/figures.zip) suggesting that MSCs/LentiVIP have the capacity to migrate into the inflamed CNS. However, the spleen and the liver were the preferred target organs for MSCs (Fig. S5; http://www.genyos.es/resources/figures.zip). No difference between MSCs/LentiGFP and nontransduced MSCs were found in the in vivo experiments, prompting us to use nontransduced MSCs as a control for the further mechanistic studies in vivo.

Intraperitoneal administration of MSCs/LentiVIP stops progression and reduces severity of EAE when administered at the peak of disease. (a) Therapeutic effect of intraperitoneal administration of MSCs/LentiVIP. MOG35–55 immunized mice were scored and stratified into four groups (untreated, MSCs, MSCs/LentiGFP and MSCs/LentiVIP) of 10 animals per group with an average clinical score of 3 ± 0.36. At this point 0.2 ml of PBS (□) or 1 × 106 MSCs (■), MSCs/LentiGFP (Δ), and MSCs/LentiVIP (▲) were inoculated intraperitoneally. After administration of the cells, the EAE clinical score were followed for over 50 days. Values are the mean ± SD of 10 mice/group. (b) Scatter plot showing the EAE score of individual mice and the median for each group at day 5, 25, and 50 posttreatment. MSC/LentiVIP treatment was significantly more efficient in reducing EAE symptoms compared to MSCs and MSCs/LentiGFP at day 25 (*p < 0.05) and day 50 (**p < 0.01) as determined by the Kruskal–Wallis test. (c) MSC/LentiVIP administration reduces MOG35 55-specific T-cell responses in draining lymph nodes (DLNs). Proliferation data are related to untreated controls. Cells were isolated from mice 7 days posttreatment and restimulated with MOG35–55 (50 μg/ml) or anti-CD3 (1 μg/ml). Values are expressed as mean ± SD of 9 mice/group, *p < 0.05. (d) Graph showing percentage of CD4+Foxp3+ Tregs in spleen (left) and DLNs (LN, right) of untreated EAE mice (white bars), MSC-treated (hatched bars) or MSC/LentiVIP-treated mice (black bars). The percentages of CD4+Foxp3+ were analyzed by flow cytometry as discussed in Materials and Methods section. Values are the mean ± SD of 8 mice/group.
Summary of MSC/LentiVIP Therapeutic Efficiency Compared to MSCs and MSCs/LentiGFP

EAE clinical scores were separated into mild (1–2.5), moderate (3–3.5), and severe (4–6). Day 0, day of treatment; day 50, days after treatment. At day 0 EAE scores ranked from 2.5 to 4. All groups (untreated, MSCs, MSCs/LentiGFP, and MSCs/LentiVIP) had similar average score at day 0. CDI is equal to the sum of the daily disease scores from each animal over 50 days. Data are mean ± SD (n = 10). LentiGFP, green fluorescent protein lentiviral vector.
MSCs/LentiVIP Significantly Reduce MOG35–55-Specific T-Cell Proliferation But Do Not Induce Foxp3+ Tregs
An important event in EAE and MS is the activation and expansion of autoreactive T-cells secreting either IFN-γ (Th1) or IL-17 (Th17) in peripheral lymphoid organs followed by their infiltration into the brain. Thus, to find an explanation for the improved therapeutic effect of MSCs/LentiVIP compared to nontransduced MSCs we set out to analyze the MOG35–55 specific recall response in DLNs of immunized mice.
We found that MOG35–55-specific proliferation of lymph node (LN) cells from MSC/LentiVIP treated mice was significantly lower compared to MSC-treated or untreated control mice suggesting that the secretion of VIP increases the immunosuppressive capacity of MSCs in vivo (Fig. 3c, left). The effect was antigen-specific since activation of total T-cells with anti-CD3 resulted in similar proliferation in all three groups (Fig. 3c, right).
MSCs have been shown to induce or expand the population of Tregs expressing Foxp3 in models of allergy-driven airway inflammation and arthritis (23,35). However, the case is less clear in EAE with only a few studies analyzing the frequency of these cells with contradictory results (39,62). In contrast, VIP has been demonstrated to induce Foxp3 expressing T-cells with immunosuppressive capacity in many models of autoimmune disease (5,17,25,32,50). We thus assessed the possible induction of Foxp3-expressing T-cells in response to MSCs or MSCs/LentiVIP in EAE. As can be seen in Figure 3d, we did not observe a significant increase in CD4+Foxp3+ T-cells in either DLNs or spleen. Apart from the Foxp3+ Tregs, there exist several Foxp3– T-cell populations with regulatory functions that suppress T-cell responses through the secretion of IL-10 (1). However, we did not detect increased levels of IL-10 or IL-4 in ex vivo cultures of splenocytes or cells from DLNs stimulated with MOG35–55, suggesting that MSCs/LentiVIP did not induce IL-10-producing Tregs or a skewing toward a Th2 response (data not shown).
MSC/LentiVIP Administration Reduced Inflammation, Increased Expression of Neuroprotective Factors, and Reduced Neuronal Degeneration in the Inflamed CNS
In addition to autoreactive T-cells, monocytes enter the CNS where they, along with activated astrocytes and microglia, secrete high levels of proinflammatory cytokines and nitric oxide (NO) resulting in axonal damage and neuronal degeneration. In the progressive stage, the innate immunity predominates, and any treatment for the chronic stage of MS needs to inactivate the diffuse inflammation that drives the axonal degeneration and subsequent neuronal cell death. We therefore studied the effect of MSCs/LentiVIP on CNS inflammation and on neurodegeneration. EAE mice were injected with either nontransduced MSCs or with MSCs/LentiVIP at the peak of disease and 7 days later killed to obtain brain and spinal cord samples. The different samples were analyzed by real-time quantitative PCR (Q-PCR) and immunohistochemistry for expression of markers of inflammation (IL-17, iNOS, TNF-α, IL-6, GFAP, and CD11b), immune regulation (IL-10, Foxp3), neuroprotection (ADNP, BDNF), and neurodegeneration (β-amyloid, β-tubulin). We found a significant decrease in the transcript levels of the proinflammatory cytokines TNF-α, IL-6, and iNOS in the spinal cords of both MSC-and MSC/LentiVIP-treated mice (Fig. 4). In contrast, IL-10 was increased in both groups compared to untreated mice, although no significance was reached. In addition, we observed increased Foxp3 mRNA in the MSCs/LentiVIP group compared with the MSCs and untreated groups, but the differences were not significant.
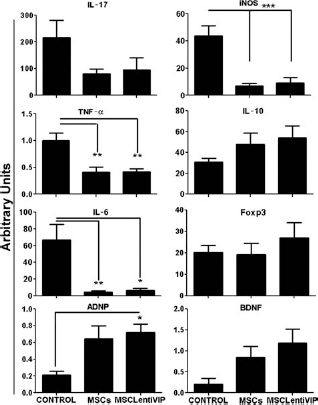
MSC- and MSC/LentiVIP-treated mice have reduced levels of proinflammatory cytokines and increased levels of neuroprotective factors in the CNS. Real-time Q-PCRs were performed on RNA extracted from spinal cords of untreated mice (control, G3.5), MSC-treated (MSCs), and MSC/LentiVIP-treated mice (MSC/LentiVIP) that were killed 7 days after cell administration. Expression levels of proinflammatory [interleukin-17 (IL-17), tumor necrosis factor (TNF)-α, IL-6] and anti-inflammatory cytokines (IL-10), Tregs [forkhead box p3 positive (Foxp3+)], oxidative burst [inducible nitric oxide synthase (iNOS)], and neuroprotective factors [activity-dependent neuroprotective protein (ADNP) and brain-derived neurotrophic factor (BDNF)] were determined for all groups as described in Materials and Methods. Results are shown as arbitrary units relative to their basal expression levels in nonimmunized littermates and shown as mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001).
VIP has been shown to induce the secretion of the neuroprotective factors BDNF-1, −3, −5 (54) and ADNP by astrocytes (2). However, we observed significant increases in BDNF and ADNP mRNA levels in both MSC- and MSC/LentiVIP-treated mice (Fig. 4). Although the Q-PCR data indicate a role of lowering inflammation and increasing neuroprotection in the therapeutic effect of MSC/LentiVIP administration, it does not reveal the differential mechanism that could explain the increased therapeutic benefits of MSCs/LentiVIP over MSCs.
In contrast, immunohistochemical analysis of CNS tissue from control, MSC-treated, or MSC/LentiVIP-treated mice showed that MSCs/LentiVIP were more efficient compared to nontransduced MSCs in decreasing both astrogliosis (GFAP) and the number of macrophages/activated microglia in the parenchyma (CD11b) (Fig. 5a, b). Immunohistochemical analysis of β-amyloid protein expression, which is a marker for neuronal degeneration, showed that MSCs/LentiVIP, but not non-transduced MSCs, significantly decreased β-amyloid expression in the inflamed brain. This correlated with significantly higher levels of the neuronal marker β-tubulin in the MSC/LentiVIP-treated group compared with the untreated or MSC-treated groups. These results were confirmed by the analysis performed at day 50 after treatment. At this stage, MSC/LentiVIP-treated mice showed higher levels of β-tubulin, increased myelin integrity (by MBP staining), and lower numbers of β-amyloid bodies compared with MSC-treated or untreated mice (Fig. S6; http://www.genyo.es/resources/figures.zip). Taken together, these data suggest that, although both MSC and MSC/LentiVIP treatment decrease CNS levels of proinflammatory cytokines (Fig. 4), MSCs/LentiVIP are more efficient in limiting the immune-mediated damage to the CNS (Fig. 5).
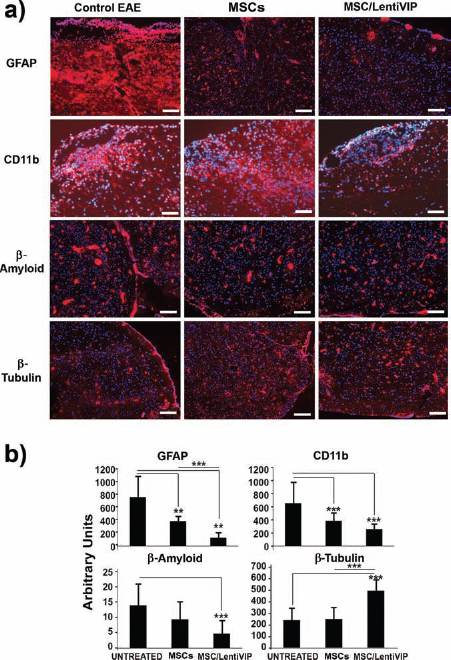
MSC/LentiVIP administration reduces astrogliosis and neurodegeneration in the CNS of EAE mice. EAE mice with a clinical score of 3–3.5 were treated with MSCs or MSCs/LentiVIP or left untreated (control EAE). Seven days later, mice were killed and the brains were embedded in OCT resin. The brains were sectioned and stained for markers of astrogliosis (GFAP), macrophage/microglia (CD11b), neuronal degeneration (β-amyloid), and neuronal markers (β-tubulin) as described in Materials and Methods. All samples were simultaneously stained with DAPI (blue) to visualize the cell nucleus. (a) Representative images of the untreated (left column), MSC-treated (center column), and MSC/LentiVIP-treated groups (right column) stained for the different markers (labels at the left) with phycoerythrin (PE) in red. Photographs were taken using a 4x objective. Scale bars: 0.2 mm. (b) Quantification of the signals obtained with the different markers (see Materials and Methods) (*p < 0.05, **p < 0.01, ***p < 0.001).
Discussion
In the present manuscript, we have developed a new gene cell therapy strategy for chronic EAE that combines the therapeutic benefits of VIP and MSCs. We have previously reported that a single injection of LentiVIP in a mouse model of severe RA resulted in a systemic expression of LentiVIP (spleen, lung, DLNs, and joints) and provided a highly effective treatment with complete regression of established disease associated with reduction of the autoimmune and inflammatory responses (13). However, in the present study, we have shown that the same strategy had no effect on established disease in a model of chronic EAE. The reason for this discrepancy could be twofold. First, in the RA model, we observed LentiVIP expression both in peripheral lymphoid organs and at the site of inflammation and tissue destruction, that is, the joints, whereas in the EAE model we could not detect VIP expression in the CNS, the target of the autoimmune attack. Second, in the RA model LentiVIP increased the number of Foxp3+ Tregs both in DLNs and in the joints whereas in the EAE model we could not detect an increase in Foxp3+ Tregs (data not shown). This suggests that, although the LentiVIP particles do reach peripheral lymphoid organs, the treatment fails because the virus particles cannot reach the inflamed CNS. It is of interest to determine whether other routes of vector administration could render better results. Indeed some authors have found that intrathecal administration of Herpes simplex virus (HSV)-based vectors expressing fibroblast growth factor (FGF), IL-4, IL-10, or interleukin-1 receptor antagonist (IL-1Ra) have some therapeutic potential (20,47,55). We therefore performed some preliminary experiments inoculating LentiGFP and LentiVIP intrathecally into EAE mice without any success due to the high mortality of the inoculated mice (data not shown).
Thus, new therapies for chronic MS must attempt not only the inhibition of the peripheral autoimmune response against CNS antigens but also stop the ongoing CNS inflammation and induce remyelination and/or neuroprotection of damaged neurons. Recently, MSCs have emerged as a promising tool for the delivery of therapeutic molecules to the CNS for several reasons (56,59). First, MSCs have been shown to home to secondary lymphoid organ and to sites of tissue injury where they inhibit inflammation/immune responses and might contribute to tissue regeneration (8,61). Second, administration of MSCs per se has been shown to infiltrate the CNS and ameliorate chronic and relapsing–remitting EAE (8). Third, MSCs can easily be isolated from different sources such as bone marrow, cord blood, or adipose tissue and expanded in vitro. We therefore used MSCs, transduced with LentiVIP (MSCs/LentiVIP), with the aims to (1) achieve VIP expression in the damaged CNS and (2) implement the MSC's own immunoregulatory and neuroprotective properties, thereby reinforcing the therapeutic potential of both agents on chronic EAE/MS.
We first showed that transduction of MSCs with LentiVIP resulted in a high and sustained expression of stable fully processed VIP, which did not significantly affect the biological properties of MSCs, including their ability to suppress T-cell responses in vitro. In order to demonstrate that the MSCs/LentiVIP secreted the fully processed VIP peptide (3.3 kDa), we combined ELISA, high-performance gel filtration, and Western blot techniques. We obtained contradictory results in terms of the VIP forms that were secreted depending on the methodology used. Western blot seemed to indicate that preproVIP (19.2 kDa) was the main form of VIP secreted while a combination of gel filtration and ELISA indicated that the MSCs/LentiVIP mostly secrete the fully processed 3.3 kDa VIP (Fig. 2a). These discrepancies could be the result of using different polyclonal antibodies for each technique. Another factor that could account for these contradictory data is the different conformational states of the proteins detected in Western blot (denatured) and ELISA (native). It is possible that the polyclonal Ab used in the ELISA kit has full access to all epitopes of 3.3-kDa VIP in its native conformation; however, several of these epitopes could be partially or completely masked in larger preproVIP. An important conclusion of these experiments is that the ELISA kit from Phoenix Pharmaceutical is highly specific for the 3.3-kDa VIP as indicated by the poor reactivity detected in the fractions containing preproVIP (in spite of it being the most abundant VIP form).
We then studied the therapeutic effect of intraperitoneal MSC/LentiVIP administration in chronic EAE. Contrary to direct injection of LentiVIP particles, systemic injection of MSCs/LentiVIP to mice with severe EAE (complete paralysis of back legs and partial paralysis of front legs) significantly reduced EAE symptoms. Treated mice regained their ability to walk with their hind legs although they still exhibited partial paresis. In contrast to direct injection of LentiVIP, administration of MSCs/LentiVIP resulted in the detection of LentiVIP transcripts in the CNS. In our hands, neither MSCs nor MSCs/LentiGFP reached any significant therapeutic activity at this stage of the disease suggesting that the combination of the biological effects of both VIP and MSCs are necessary to achieve therapeutic benefits in chronic disease.
The innate immune response predominates in chronic MS and T-cell infiltration into the CNS gray and white matter is low in both primary and secondary progressive MS. However, larger numbers of T-cells have been found in the meninges where they produce IFN-γ and are thought to interact with inflammatory macrophages (3). In addition, IL-17 transcripts are upregulated in chronic MS lesions (40) highlighting the involvement of T-cells in chronic MS. Functional studies demonstrated that MSC/LentiVIP administration was more potent compared to MSCs alone in inhibiting the in vivo antigen-specific T-cell autoimmune responses in peripheral LNs. Both MSCs (64) and VIP (7) have been shown to induce DCs with lower expression of costimulatory molecules that are defective in inducing T-cell responses. Importantly, VIP can directly induce cell cycle arrest at multiple levels and anergy in activated human T-cells (2,50). Thus, we suspect that MSCs/LentiVIP could induce a state of unresponsiveness/anergy in T-cells both during their initial activation in the LNs and also in activated T-cells present in the periphery or in the CNS.
The inhibition of the T-cell response did not correlate with increased numbers of Foxp3+ Tregs nor higher IL-10 expression levels in DLNs or spleens in MSC/LentiVIP-treated mice compared to MSCs alone. The role of Foxp3+ Tregs is MS is not well known. Tregs appear to have impaired function in RRMS, and they are not readily detected in MS lesions (30). Whether or not MSCs induce functional Tregs in EAE is also not clear (62). Our results showed that MSCs/LentiVIP did not increase the frequency of CD4+Foxp3+ T-cells in the DLNs, despite the expression of VIP. We did detect a small increase in Foxp3 mRNA in the CNS of mice treated with MSCs/LentiVIP compared with those treated with MSCs only but these differences were not significant.
In the CNS both MSCs/LentiVIP and MSCs significantly inhibited the expression of proinflammatory cytokines and iNOS compared to control mice. However, at day 50, myelin immunoreactivity in the CNS from MSC/LentiVIP-treated mice was significantly higher compared to MSC-treated or control mice showing that MSCs/LentiVIP were more efficient in inhibiting the demyelinating immune attack and/or increasing remyelination in the CNS. Brain tissue sections from MSC/LentiVIP-treated mice showed lower GFAP (a marker of astrocyte activation) staining compared to both MSC- or PBS-treated mice. The reduction in astrocyte activation could be the consequence of the reduced neuronal cell death in MSC/LentiVIP-treated mice, which was evidenced by the lower accumulation of β-amyloid deposits and higher levels of β-tubulin immunoreactivity in the CNS. The decrease in neurodegeneration could also be explained by the potent neuroprotective effects of VIP through the inactivation of microglia (11) or modulation of astrocyte activity (9). VIP induces BDNF and ADNP, which are growth factors involved in neuroprotection (54); therefore, it is reasonable to hypothesize that the enhanced effect observed with MSCs/LentiVIP could be due to enhanced BDNF and/or ADNP production. However, we observed similar BDNF and ADNP increments in MSC/LentiVIP- and MSC-treated mice, although the levels were increased in both groups compared to untreated controls. Although the specific mechanism explaining the MSC/LentiVIP-mediated effects remains elusive, we propose the following events as instrumental in achieving the therapeutic effect observed by intraperitoneal administration of MSCs/LentiVIP: (1) the increased inhibition of autoimmune T-cells, (2) the reduction in astrogliosis that results in a reduced infiltration of CD11b+ cells, and (3) the increased neuronal survival.
Our data indicates that, although MSCs possess the capacity to actively migrate into the inflamed CNS, only a fraction of systemically injected cells reached the target while most of the engrafted cells ended in the spleen and liver. This has also been observed by other groups (61,62). In order to increase the amount of MSCs that reach the target areas, some groups have developed different strategies to deliver MSCs directly to the damaged CNS. Intracerebroventricular (ICV) transplantation and intrathecal injection of MSCs have been shown to be safe in MS mouse models and human MS patients (33,34). However, in a recent study by Grigoriadis et al. (30), BM-MSCs were ICV transplanted into mice with mild and severe chronic EAE in C57Bl/6 mice. Whereas ICV transplanted BM-MSCs ameliorated mild EAE, they did not have any effect on severe EAE. In addition, ICV transplantation of BM-MSCs into severe EAE resulted information of fibrotic cell masses in the ventricles infiltrated by lymphocytes. Further studies are needed to assess whether the possible benefits of ICV transplantation or intrathecal injection of MSCs in MS are worth the risks it may bring, considering that MS is not a life-threatening disease. Many factors will decide which route should be used as well as the (i) number of cells, (ii) disease severity and stage, and (iii) safety. Maybe a combination of both IV and intrathecal administration of MSCs could synergistically increase the therapeutic efficacy of MSCs by simultaneously inhibiting the peripheral immune response and increasing the local immunomodulation and neuroprotection in the CNS.
VIP has a wide variety of biological activities such as smooth muscle relaxation, secretion of water in the intestine, circadian timekeeping, vasodilation, etc. The potential side effects of constitutive overexpression are therefore a matter of concern. However, we did not detect any signs of adverse effects such as watery diarrhea or vasodilation/hypotension in our treated animals. We think that the amounts of VIP secreted by the injected cells are not sufficient to cause these symptoms. However, we cannot rule out other potential side effects in a longer period of time. It is therefore evident that MSCs expressing VIP constitutively are not optimal for its translation into clinic. We are therefore developing doxycycline-induced MSCs/LentiVIP to increase safety (work currently going in the laboratory).
In summary, we have demonstrated the therapeutic potential of gene cell-based strategies for treatment of chronic EAE, an animal model for MS. In particular we have demonstrated that MSCs can be used to deliver VIP and that this combination is far more efficient than the use of MSCs or lentiviral vectors expressing VIP alone. The easy growth of MSCs and the integration of lentiviral vectors allow the design of easy protocols to scale up the number of MSCs/LentiVIP for the potential application in human patients. In addition, the possibility of using allogeneic MSCs opens up the possibility of banking gene-modified cells for in detail analyses before clinical use.
Footnotes
Acknowledgments
This work has been supported by the Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (ISCIII), and Fondo Europeo de Desarrollo Regional (FEDER) from the European Union, through the resesarch project No. PS09/00340. We also acknowledge financial support by the Consejería de Salud and the Consejería de Innovación Ciencia y Empresa from the Junta de Andalucia and FEDER/Fondo de Chesión Europeo (FSE) through the Programa Operativo FEDER/FSE de Andalucia 2007-2013 and projects No. P09-CTS-04532, No. PI0001/2009 and PAIDI-Bio-326. M.C. and F.M. are financed by Fundación Progreso y Salud (Consejería de Salud - Junta de Andalucía). K.B and M.G.T. are financed by P09-CTS-04532 and PI0001/2009 grants, respectively. P.A. and P.M. have a Miguel Servet (CP09/00228) and Sara Borrell contracts, respectively [Fondo de Investigaciones Sanitarias (FIS) - Institute of health Carlos III]. The authors declare no conflicts of interest.