
Abstract
Mesenchymal stem cells (MSCs) are efficacious in a variety of intractable diseases. While bone marrow (BM)-derived MSCs (BM-MSCs) have been widely investigated, MSCs from other tissue sources have also been shown to be effective in several autoimmune and inflammatory disorders. In the present study, we simultaneously assessed the therapeutic efficacy of human BM-MSCs, as well as MSCs isolated from adipose tissue (Ad-MSCs) and umbilical cord Wharton's jelly (UC-MSCs), in experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis (MS). Prior to in vivo experiments, we characterized the phenotype and function of all three MSC types. We show that BM-MSCs were more efficient at suppressing the in vitro proliferation of mitogen or antigen-stimulated T-cell responses compared to Ad-MSCs and UC-MSCs. Notably BM-MSCs induced the differential expression of cytokines from normal and stimulated T-cells. Paradoxically, intravenous transplantation of BM-MSCs into C57Bl/6 mice with chronic progressive EAE had a negligible effect on the disease course, even when multiple MSC injections were administered over a number of time points. In contrast, Ad-MSCs had the most significant impact on clinical and pathological disease outcomes in chronic progressive and relapsing–remitting EAE models. In vivo tracking studies revealed that Ad-MSCs were able to migrate to the central nervous system (CNS), a property that most likely correlated with their broader expression of homing molecules, while BM-MSCs were not detected in this anatomic region. Collectively, this comparative investigation demonstrates that transplanted Ad-MSCs play a significant role in tissue repair processes by virtue of their ability to suppress inflammation coupled with their enhanced ability to home to the injured CNS. Given the access and relatively ease for harvesting adipose tissue, these data further implicate Ad-MSCs as a cell therapeutic that may be used to treat MS patients.
Keywords
Introduction
Although the precise mechanisms attributed to the initiation of multiple sclerosis (MS) have yet to be clearly elucidated, current consensus suggests that the disease is maintained by autoreactive T-cells that target proteins expressed predominantly on the myelin sheath and, to a lesser extent on axons, resulting in central nervous system (CNS) tissue injury (52). Despite early attempts at repair, accumulating axonal and neuronal damage due to chronic neuroinflammation and neurodegeneration ultimately leads to increasing and permanent neurological dysfunction (24). A number of therapeutic approaches have been targeted towards the immune component of the disease process. While these have been shown to alter the disease course, they are, however, only partially effective and have little impact on disease progression (41). Thus, there is a clear need to find new therapeutic approaches to treat this severely debilitating disease. Stem cell therapies, including those based on transplantation of multipotent stromal cells [also known as mesenchymal stem cells (MSCs)], have the potential to act as immunomodulatory as well as neuroprotective agents and therefore represent a promising novel cell therapeutic for MS.
MSCs were originally identified from the bone marrow (BM), where they represent only a minor fraction of the total number of BM cells (58). Although the in vivo identity of MSCs is still under debate (16,54), these cells play an important role in supporting hematopoiesis and maintaining homeostasis within the BM niche (74). In addition to the relative ease in which these cells can be isolated and expanded ex vivo, MSCs possess a number of functional properties pertinent in a therapeutic transplantation setting. Extensive investigations into their immunomodulatory properties have shown that MSCs can inhibit T-cell (22,43), B-cell (15), and antigen-presenting cell (8) function; are protected from cytotoxic T-cell-mediated lysis (63); and can modulate the actions of T-regulatory cells (Tregs) (21). Systemically infused MSCs preferentially home to inflamed and injured tissues (38) and induce clinical recovery via a number of mechanisms, such as the suppression of inflammatory responses, exertion of potent antioxidant effects, and the secretion of trophic factors capable of stimulating endogenous repair pathways (68). Further corroborating these observations, MSC transplantation can be effective at reducing inflammatory responses in experimental animal models of myocardial infarction (47), diabetes (26), rheumatoid arthritis (1), and lupus nephritis (11).
The potential benefits of MSC treatment in MS have been highlighted by a series of transplantation studies in experimental autoimmune encephalomyelitis (EAE). Two early reports demonstrated that systemic transplantation of BM-MSCs can modulate immune responses to induce peripheral tolerance and ameliorate disease (29,75). These peripheral effects have since been confirmed in other studies following intravenous (IV) and intraperitoneal (IP) delivery of MSCs (3,14,31,61,62). While several groups report that a small number of MSCs of either mouse (14,39) or human origin (3,32) are capable of homing to the CNS to exert in situ immunomodulatory and neuroprotective effects, there is little evidence for their long-term engraftment.
Since their identification within the BM compartment, MSCs have been isolated from almost every tissue in the body (18). Despite their similarities, comparative analyses on tissue-specific MSCs have revealed differences that may be relevant to their ability to modulate immune responses (9,37). To date, a comparative study of the potential of tissue-specific MSCs to inhibit autoimmune-mediated demyelination has not yet been carried out. Here, we report on the characterization of human MSCs isolated from three different tissues, BM, adipose tissue (Ad), and umbilical cord Wharton's jelly (UC), with a particular focus on comparing their immunomodulatory effects in vitro. The therapeutic efficacy of these three MSC populations was then evaluated using recombinant myelin oligodendrocyte glycoprotein (rMOG)-induced EAE, a model of MS in which both T- and B-cells contribute to disease pathogenesis.
Materials and Methods
Mice
C57Bl/6 mice were purchased from Monash University Animal Services. Nonobese diabetic (NOD)/Lt mice were purchased from the Walter and Eliza Hall Institute animal breeding facility (Melbourne, Australia). MOG T-cell receptor (TCR) transgenic (2D2) mice and macrophage colony-stimulating factor receptor (Cfms)-green fluorescent protein (GFP) mice were bred at the Monash University Animal Services breeding facility. All mice were bred and maintained under pathogen-free conditions.
Mesenchymal Stem Cell Culture
Normal or enhanced green fluorescent protein (e)GFP-expressing human BM-MSCs purchased from the Tulane Center for Stem Cell Research and Regenerative Medicine (Tulane University, New Orleans, LA, USA) were cultured in a medium consisting of αMEM supplemented with 16.5% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen, Carlsbad, CA, USA). Cells were seeded at a density of 60 cells/cm2 in tissue culture flasks (BD Biosciences, Franklin Lakes, NJ, USA), and medium was changed every 3–4 days. BM-MSCs were passaged upon reaching ~80% confluence (typically after 10-12 days of culture) using 0.25% trypsin-EDTA (Invitrogen). Human Ad-MSCs and UC-MSCs purchased from ScienCell (Carlsbad, CA, USA) were cultured in medium consisting of αMEM containing 5% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1% MSC growth supplement (ScienCell). Cells were seeded in a flask precoated with 2 μg/cm2 poly-l-lysine (Sigma-Aldrich, St. Louis, MO, USA) at a density of 5,000 cells/cm2 and passaged when they reached 90% confluence (typically after 3 days) using 0.25% trypsin-EDTA. For some in vitro experiments, MSCs were cultured in the presence of human recombinant interferon (IFN)-γ (R&D Systems, Minneapolis, MN, USA) at 10 ng/ml for the final 72 h of culture.
Flow Cytometry
Phenotypic analysis by single color flow cytometry was performed by staining 0.5 × 106 cells with primary antibodies for 20 min at 4°C. Where applicable, cells were then stained with a goat anti-mouse or goat anti-rat Alexa Fluor 647 secondary antibody for 20 min at 4°C. The relevant isotype control antibody was used as a negative control. Data acquisition was performed using a FACSCalibur Flow Cytometer (BD Biosciences), and data were analyzed using Gatelogic Software (Inivai Technologies, Mentone, Victoria, Australia). Primary antibodies were purchased from BD Biosciences, and secondary antibodies were purchased from Invitrogen. Cell surface expression of adhesion molecules was assessed using a Human Cell Surface Marker Screening Panel test kit kindly provided by BD Biosciences.
Gene Expression Analysis
Total RNA was extracted from 1 × 106 MSCs at passages 3 and 5, using a High Pure RNA Isolation Kit (Roche, Indianapolis, IN, USA) as per the manufacturer's instructions. cDNA was prepared from 0.5 μg of total RNA using a SuperScript III First-Strand Synthesis System RT-PCR Kit and oligo (dT) primers (Invitrogen). PCR was performed using Taq DNA Polymerase (Invitrogen) with 1 μl cDNA and primer pairs for human chemokine receptors (10,28) under the following cycling conditions: 94°C for 5 min and then 35 cycles of 94°C for 1 min, 55°C for 45 s, and 72°C for 1 min followed by 72°C for 10 min. The PCR products were resolved by electrophoresis in 2% agarose gels and the bands visualized under UV light.
EAE Induction and Cell Transplantation
EAE was induced in 10- to 12-week-old female C57Bl/6 or NOD/Lt mice with the extracellular domain of mouse recombinant (r)MOG (amino acid residues 1-117 of the mature protein) (6) as previously described (56). For MSC transplantation, cells were injected via the lateral tail vein or into the peritoneal cavity in a volume of 200 μl on the days indicated. Control mice received injections of equal volumes of phosphate-buffered saline (PBS, Invitrogen). Mice were monitored daily, and clinical scores were assigned according to an arbitrary clinical scale as described (53). Mice were humanely killed upon reaching a clinical score of 4. All animal experiments were carried out in accordance with animal ethics guidelines, as set out by the Monash University School of Biomedical Sciences Animal Ethics Committee.
T-Cell Proliferation Assays and Cytokine Production
Spleens were collected from normal C57Bl/6 mice, 2D2 mice expressing the transgenic Vα3.2/Vβ 11TCR (7), or rMOG-immunized mice. Single cell suspensions were prepared in complete Roswell Park Memorial Institute (RPMI) medium containing 10% heat-inactivated FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from Invitrogen), 50 μM 2- mercaptoethanol (Sigma-Aldrich), and 1 mM sodium pyru-vate (Sigma-Aldrich). Red blood cells were lysed in 3 mllysis buffer (155 mM NH4CL, 10 mM KHCO3, 0.1 mM EDTA, Sigma-Aldrich) for 1 min, and then splenocytes were seeded in triplicate in 96-well, flat-bottommed microtiter plates (Nunc, Roskile, Denmark) at a concentration of 2.5×105 cells per well. Splenocytes were cultured in medium alone or in the presence of either 20 μg/ml MOG35-55 (GL Biochem, Shanghai, China), 20 μg/ml rMOG, 800 ng/ml ionomycin, and 20 pg/ml phorbol myristate acetate (PMA, both from Sigma-Aldrich), or in wells precoated with 10 μg/ml anti-CD3 and 10 μg/ml anti-CD8 antibodies (both from BD Biosciences). Cells were incubated at 37°C for 72 h with the addition of 1 μCi/well [3H]thymidine (Perkin Elmer, Waltham, MA, USA) for the last 18 h of culture. Cells were harvested onto filter mats (Perkin Elmer) and incorporated radioactive nucleic acids counted using a Top Count NXT Scintillation Counter (Packard Biosciences, Meriden, CT, USA). For coculture experiments, MSCs were added at concentrations ranging from 0.002 to 0.25 × 105 cells per well prior to the addition of 2.5 × 105 splenocytes.
For analysis of cytokine production, 2.5 × 106 2D2 splenocytes were cultured in 24-well plates (BD Biosciences) for 48 h in medium alone or in the presence of 20 μg/ml MOG35-55, to a final volume of 1 ml. For coculture experiments, 0.25 × 106 MSCs, 500 μl MSC conditioned medium, or 500 μl control cell culture medium was added. Quantitative analysis of interleukin (IL)-2, IL-6, IL-10, IL-17A, IFN-γ, and tumor necrosis factor (TNF)-α was performed using a mouse cytometric bead array kit (BD Biosciences) according to the manufacturer's instructions. Acquisition of events was performed on a BD FACSCanto II Flow Cytometer and data analyzed and fitted to a four-parameter logistic equation using the FCAP Array Software (Soft Flow, St. Louis Park, MN, USA).
Anti-MOG Antibody Determination
Blood was collected at the end point of experiments by cardiac puncture and sera isolated. The antibody isotypes IgG, IgG1, IgG2a, IgG2b, IgG3, IgA, and IgM that specifically bound to rMOG were measured by ELISA. Briefly, flat-bottommed 96-well plates were coated with 100 μl of rMOG (5 μg/ml) in 0.05 M carbonate buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.6; Sigma-Aldrich) overnight at 4°C. Wells were blocked with Pierce protein-free blocking medium (Thermo Fischer Scientific, Rockford, IL, USA) supplemented with 1.5% normal goat serum for 3 h at room temperature. For specific isotype antibody responses, serum was diluted as follows: 1/8,000 for IgG and IgG1, 1/50 for IgG2a, 1/2,000 for IgG2b, 1/200 for IgG3, 1/50 for IgA, and 1/100 for IgM. Serum from naive C57BL/6 mice was used as a negative control and the anti-MOG monoclonal antibody clone 8-18C5 (produced in-house by hybridoma) diluted to 5 μg/ml was used as a positive control. Antibody binding was revealed using horseradish peroxidase-conjugated goat anti-mouse isotype antibodies (Caltag Laboratories, Burlingame, CA, USA) diluted 1/2,000 or a polyvalent IgG,A,M antibody (Sigma-Aldrich) diluted 1/1,000. O-Phenylenediamine (Sigma-Aldrich) was used as a substrate and the reaction performed according to the manufacturer's protocol. Optical density (OD) was measured at 492 nm on a Benchmark Plus Microplate Spectrophotometer (BioRad, Hercules, CA, USA).
Histological Analysis of CNS Tissue
For histological analysis of CNS tissue, the brain and spinal cord were dissected from mice and fixed in 10% formalin (Sigma-Aldrich). Sections (5 μm) were cut from paraffin-embedded tissue and stained with hematoxylin– eosin, luxol fast blue, and Bielschowsky silver impregnation to assess inflammation, demyelination, and axonal damage, respectively. Reagents for histological staining were purchased from Grale Scientific (Ringwood, Victoria, Australia).
Lentiviral Transduction of MSCs
The parental pWPI lentiviral plasmid, a second generation, self-inactivating, bicistronic lentiviral vector was obtained from Professor Didier Trono (Ecole Polytechnique Federale de Lausanne). This vector backbone incorporates an elongation factor-1α promoter, an internal ribosomal entry site (IRES)-enhanced green fluorescent protein (eGFP) cassette, and the Woodchuck posttranslation regulatory element. Using standard molecular biology techniques, the cDNA for firefly luciferase (fluc) was subcloned and blunt-end ligated upstream of the IRES-eGFP cassette generating the transfer vector pWPI-fluc-IRES-eGFP. Viral stocks were generated by transfecting pWPI-fluc-IRES-eGFP together with pSPAX2 and pMD.2G using Fugene6 (Roche) into 293T cells (ATCC CRL-11268; Manassas, VA, USA) as previously described (67). Typically, 0.5–1×107 transducing units/ml of unconcentrated vector was generated in this manner. Nonmodified MSCs were transduced twice at a multiplicity of infection of 50 over a 48-h period with unconcentrated lentiviral supernatant diluted 1:2 with culture medium in the presence of 8 μg/ml protamine sulfate (Sigma-Aldrich). Transduction medium was replaced with fresh medium 16 h after each transduction.
Bioluminescence Imaging
Animals were transplanted via the lateral tail vein with 1×106 transduced MSCs. To image gene-modified cells, animals were injected IP with 200 μl d-luciferin (15 mg/ml in PBS, VivoGlo Luciferin, Promega, San Luis Obispo, CA, USA) on days 0, 2, 7, 14, 21, and 28 post-cell injection. On these days, mice were anesthetized for 10 min with 2.5% isoflurane prior to bioluminescence imaging, which was performed with an IVIS 200 system (Xenogen, Alameda, CA, USA). The fluc luminescent signal intensity was analyzed using Living Image 3.2 Software (Xenogen).
Tracking of GFP+ MSCs by PCR
Organs from EAE mice injected with BM-MSCs or Ad-MSCs overexpressing eGFP were dissected and genomic DNA extracted using a Gentra Puregene Tissue Kit (Qiagen, Venlo, Netherlands) according to the manufacturer's instructions. PCR for eGFP was performed with Platinum Pfx DNA Polymerase (Invitrogen) and 100 ng of cDNA under the following cycling conditions: 94°C for 5 min and then 42 cycles of 94°C for 30 s, 64.9°C for 30 s, and 68°C for 30 s followed by 68°C for 10 min. PCR for the housekeeping gene β-actin was performed with Platinum Taq DNA Polymerase (Invitrogen) and 100 ng of cDNA under the following cycling conditions: 94°C for 2 min and then 32 cycles of 94°C for 30 s, 53°C for 30 s, and 72°C for 1 min followed by 72°C for 10 min. Primer pairs were eGFP: 5′-TACGGCAAGCTGACCCTGAAGTTC-3′, 5′-CGTCGTCCTTGAAGAAGATGGTGCG-3′; β-actin: 5′-GGCACCACACCTTCTACAATG-3′, 5′-CTGCTGC TGAAGCTGTAG-3′. The PCR products were resolved by electrophoresis in 2% agarose gels, and the bands were visualized under UV light. DNA from cFMS-GFP mice and uninjected mice were positive control (+ve) and negative controls, respectively.
Statistical Analysis
Data are presented as the mean ± standard error of the mean (SEM). All statistical analyses were performed using Prism 5.04 (Graphpad Software, San Diego, CA, USA). Significant differences between two groups were determined using an unpaired Student's t test. Statistical analysis of three or more groups was performed using one-way ANOVA with Kruskal–Wallis and Dunn's post hoc test. Values of p < 0.05 were considered to be significant.
Results
Immunophenotype of MSCs
Flow cytometric analysis was used to examine the expression of cell surface markers by MSCs. Consistent with the minimal criteria proposed by Dominici et al. (23), BM-MSCs, Ad-MSCs, and UC-MSCs expressed the canonical MSC antigens CD73, CD90, and CD105 but lacked expression of the hematopoietic markers CD45, CD34, CD11b, and CD19 (data not shown). HLA-DR (MHC class II) was not constitutively expressed but could be induced by IFN-γ stimulation (Fig. 1). All three MSC types expressed HLA-ABC (MHC class I) but lacked expression of the costimulatory molecules B7-H2, CD28, CD40, CD80, and CD86. Notably CD47, which interacts with signal regulatory protein a (SIRPa) to negatively regulate phagocytosis (51), was highly expressed on all three MSC types.

Phenotypic profile of MSCs. Mesenchymal stem cells (MSCs) from bone marrow (BM), adipose tissue (Ad), and umbilical cord (UC) were cultured in the absence or presence of 10 ng/ml interferon (IFN)-γ for 72 h, and cell surface expression of human leukocyte antigen (HLA)-DR was examined by flow cytometry. Cell surface expression of other antigens was examined under standard culture conditions. Representative histograms of two independent experiments are shown in gray. Isotype controls are indicated by the black curve. B7-H2, inducible T-cell costimulator ligand; CD, cluster of differentiation.
BM-MSCs Suppress T-Cell Proliferation More Efficiently Than Ad-MSCs and UC-MSCs
The immunomodulatory effects exerted by MSCs represent an important mechanism by which these cells can mediate clinical recovery in experimental animal models (68). Given the central role of T-cells in EAE pathogenesis (52), we investigated the immunosuppressive potential of MSCs by simultaneously comparing their ability to inhibit T-cell proliferative responses under various in vitro conditions. Splenocytes from C57Bl/6 mice stimulated with T-cell mitogens were cocultured with increasing numbers of MSCs, and then their proliferation was analyzed after 72 h by [3H]thymidine incorporation. BM-MSCs had a dramatic effect on the proliferation of PMA/ionomycin activated splenocytes, resulting in greater than 90% inhibition of T-cell proliferation for all four MSC/splenocyte ratios tested (Fig. 2A). A dose-dependent effect was observed following the addition of either Ad-MSCs or UC-MSCs, as demonstrated by decreasing T-cell proliferation with increasing concentrations of MSCs. At the lowest ratio of 1:1250, BM-MSCs suppressed T-cell proliferation by at least 90%, while the relative suppression by Ad-MSCs and UC-MSCs was only 47% and 22%, respectively. Congruent with these observations, BM-MSCs were more efficient at suppressing TCR-dependent T-cell proliferation driven by the addition of anti-/CD28 antibodies (Fig. 2B). To assess whether MSCs could also inhibit autoantigen-specific T-cell responses in vitro, we used splenocytes from 2D2 mice that express the transgenic Vα3.2/Vβ11 TCR specific for the MOG35–55 peptide in the context of I-Ab (7). MSCs suppressed the proliferation of 2D2 splenocytes stimulated with MOG35–55 in a dose-dependent manner, with BM-MSCs again inhibiting T-cell proliferative responses more efficiently compared to Ad-MSCs and UC-MSCs (Fig. 2C).

MSCs inhibit T-cell proliferation. C57Bl/6 splenocytes stimulated with either phorbol myristate acetate/ionomycin (PMA/I) (A) or anti-CD3/anti-CD28 antibodies (B) or 2D2 splenocytes stimulated with myelin oligodendrocyte glycoprotein (MOG)35–55 (C) were cultured in the presence of increasing doses of MSCs (expressed as MSC:splenocyte ratio). Proliferative responses were measured by [3H]thymidine incorporation and expressed as the mean counts per minute (CPM) + SEM of two independent experiments (n = 6 mice). *p < 0.05, **p < 0.01, ***p < 0.001 versus splenocytes without MSCs.
Soluble Factors Secreted by BM-MSCs Modulate Cytokine Production
To assess whether MSCs could modulate the production of cytokines that are relevant to EAE pathogenesis, supernatants were collected from cocultures of MSCs and MOG35–55-stimulated 2D2 splenocytes and cytokines quantified by cytometric bead array. Splenocytes cultured in the presence of MSCs produced significantly lower amounts of the Th1 cytokines IFN-γ and IL-2 (Fig. 3A). Secretion of IL-17A was also significantly inhibited by BM-MSCs and Ad-MSCs; however, there was no change in IL-17A secretion when splenocytes were cocultured with UC-MSCs. Interestingly, while the presence of Ad-MSCs or UC-MSCs led to a reduction in TNF-α secretion, BM-MSCs had no effect on the secretion of this cytokine. Increased production of IL-6 and IL-10 was also observed when splenocytes were cocultured with BM-MSCs.

BM-MSCs modulate cytokine production by autoantigen-stimulated splenocytes. Supernatants were collected from cocultures of 2D2 splenocytes and MSCs or 2D2 splenocytes and MSC conditioned medium (CM) after 48 h, and cytokines were quantified by cytometric bead array. (A) MOG35–55-stimulated splenocytes cultured with MSCs at a ratio of 1:10. (B) Unstimulated splenocytes cultured with MSCs at a ratio of 1:10. (C) Unstimulated splenocytes cultured with CM or control culture medium (BM media or Ad/UC media). Data represent the mean ± SEM of two independent experiments (n = 5 mice). *p < 0.05, **p < 0.01, ***p < 0.001 versus splenocytes without MSCs, ^p < 0.001 versus Ad-MSC and UC-MSC. IL, interleukin; TNF, tumor necrosis factor.
Likewise, when we assessed the levels of IL-6, TNF-α, and IL-10 produced by unstimulated splenocytes, a dramatic increase in the production of these cytokines was observed in the presence of BM-MSCs only (Fig. 3B). Ad-MSCs or UC-MSCs did not elicit such a response. In addition to cell–cell contact mechanisms, MSCs can modulate immune responses through the secretion of soluble factors (22,62,71). Conditioned medium from confluent cultures of BM-MSCs was therefore added to unstimulated or MOG35–55-stimulated splenocytes to determine if the increase in TNF-α, IL-6, and IL-10 was due to the release of soluble factors. We found that production of all three cytokines was significantly increased in the presence of BM-MSC conditioned medium, but not in the presence of control culture medium or conditioned medium from cultures of Ad-MSCs or UC-MSCs (Fig. 3C).
BM-MSC Transplantation Does Not Have an Enhanced Therapeutic Effect on the Clinical Course in Chronic Progressive EAE
To compare the therapeutic effect of tissue-specific MSCs in vivo, we induced chronic progressive EAE by immunizing C57Bl/6 mice with the extracellular domain of rMOG. Three IV injections of 1×106 MSCs were then administered prior to the onset of clinical signs on days 8, 10, and 12 post-disease induction. There was an overall reduction in the daily mean clinical score of mice that received BM-MSCs, Ad-MSCs, or UC-MSCs compared to PBS injected controls; however, no difference between the different MSC-treated groups was observed (Fig. 4A). Notably, cumulative and maximal disease scores were significantly reduced in Ad-MSC- and UC-MSC-treated animals in comparison to the PBS-injected mice (Table 1). When splenocytes from EAE mice were restimulated in vitro with rMOG, the proliferative response was significantly reduced for mice receiving MSCs compared to PBS controls (Fig. 4B) and no difference between the three MSC types was found. A similar reduction in T-cell proliferative responses was also observed upon restimulation with anti-CD3/CD28 antibodies (Fig. 4C), indicating that MSC suppression was not limited to antigen-specific T-cell clones. Analysis of serum rMOG-specific antibody responses revealed no difference between MSC-treated mice and controls (Fig. 4D).
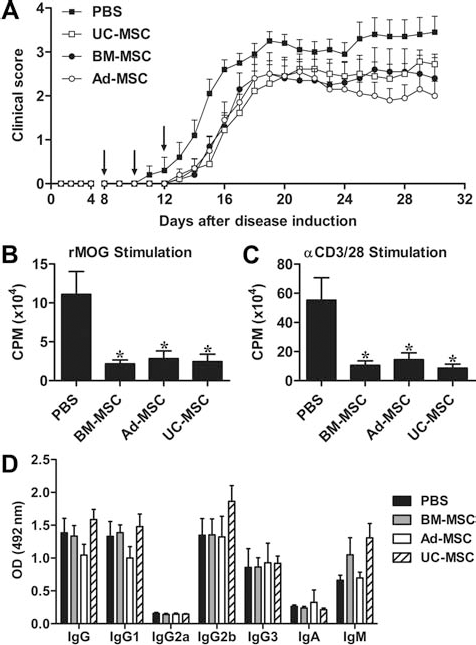
Multiple early MSC injections reduce EAE severity. C57Bl/6 mice were immunized with recombinant MOG (rMOG) and MSCs administered IV. (A) MSCs (1×106) were injected on days 8, 10, and 12 postimmunization. Arrows, days of MSC injections. Proliferative response of splenic T-cells from MSC-treated mice and controls to rMOG (B) and anti-CD3/anti-CD28 (C). (D) Serum rMOG-specific IgG, IgA, and IgM antibody responses from MSC-treated mice and controls. Data represent the mean ± SEM of two independent experiments (n = 9–10 mice). *p < 0.05 versus PBS controls.
Clinical Outcome of Chronic Progressive EAE Mice Treated With MSCs

Data represent mean ± SEM.
p < 0.05
p < 0.01 versus PBS. EAE, experimental autoimmune encephalomyelitis; MSCs, mesenchymal stem cells; PBS, phosphate-buffered saline (control); BM, bone marrow; Ad, adipose tissue; UC, umbilical cord.
To determine if MSC administration could impact on the clinical course of disease once immune-mediated damage had already been established within the CNS, 1×106 MSCs were injected after the onset of clinical symptoms on days 12, 14, and 16 post-disease induction. In contrast to the previous treatment protocol, no beneficial effect was observed when MSCs were administered at these later time points (Fig. 5A). Given that treatment prior to disease onset was more efficacious, we next assessed whether a single bolus injection of MSCs would have any beneficial effect in EAE mice. MSCs (2×106) were administered IV on day 8 post-disease induction; however, there were no differences in disease onset or severity between MSC-treated mice and controls (Fig. 5B).

MSCs have no effect when administered at disease onset or in a single dose. C57Bl/6 mice were immunized with rMOG and MSCs administered IV. (A) MSCs (1×106) were injected on days 12, 14, and 16 postimmunization. (B) MSCs (2×106) were injected on day 8 postimmunization. Data represent the mean ± SEM (n = 5 mice). Arrows, days of MSC injections.
Ad-MSC Transplantation Attenuates Relapsing–Remitting EAE
As an adjunct to the chronic progressive EAE studies, we further examined the therapeutic potential of Ad-MSCs in a relapsing–remitting model (Fig. 6). Implementing a similar treatment protocol, 1×106 Ad-MSCs or PBS were administered IV to NOD/Lt mice on days 8, 10, and 12 post-rMOG immunization. Intriguingly, Ad-MSCs had a more profound effect on the development of EAE in this animal model, with a marked reduction in the clinical and maximum scores as well as a delay in the disease onset (Fig. 6A and Table 2). Given that IP. delivery of MSCs has previously been shown to ameliorate EAE through suppressing immune responses in the periphery (31,61,62), we also assessed the effect of high-dose IP injections of Ad-MSCs. When 5×106 Ad-MSCs were delivered on days 8, 10, and 12, there was an even greater amelioration of the disease compared to administration of low-dose IV injections (Fig. 6B and Table 2). Consistent with the clinical data, hematoxylin and eosin staining of spinal cord sections showed a reduction in the inflammatory infiltration in Ad-MSC-treated mice compared to controls (Fig. 6C, F), which corresponded with a decrease in demyelination (Fig. 6D, G) and axonal damage (Fig. 6E, H).

Ad-MSC transplantation suppresses relapsing–remitting EAE. NOD/Lt mice were immunized with rMOG and MSCs administered on days 8, 10, and 12 postimmunization. (A) Ad-MSCs (1×106) were injected IV. (B) Ad-MSCs (5×106) were injected IP. Arrows, days of Ad-MSC injections. Data represent the mean ± SEM (n = 4–6 mice). Representative spinal cord sections from mice injected IP with PBS (C–E) or Ad-MSCs (F–H) were stained with hematoxylin and eosin to detect inflammatory infiltrates (C, F), luxol fast blue to detect the degree of demyelination (D, G), and Bielschowsky silver stain to detect axonal damage (E, H). Original magnification: 400×.
Clinical Outcome of Relapsing–Remitting EAE Mice Treated With Ad-MSCs

Data represent mean ± SEM.
p < 0.05
p < 0.01 versus PBS.
Potential Molecular Trafficking Mechanisms Vary Between Tissue-Specific MSCs
In order to identify any cell-specific differences that may explain the discrepancy between the in vitro and in vivo immunomodulatory effect of BM-MSCs, we first examined the expression of molecules that may influence their in vivo trafficking. Extensive profiling of cell surface adhesion molecules by flow cytometry is summarized in Table 3. These results showed that, under standard culture conditions, all three MSCs lacked expression of molecules involved in selectin-mediated cellular migration, including E-selectin (CD62E), l-selectin (CD62L), P-selectin (CD62P), and P-selectin glycoprotein ligand (PSGL)-1 (CD162). Of the immunoglobulin superfamily cell adhesion molecules, activated-leukocyte cell adhesion molecule (ALCAM; CD166) and melanoma cell adhesion molecule (MCAM; CD146) were expressed by all three MSCs; however, intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106) were expressed only by a subpopulation of Ad-MSCs and UC-MSCs. MSCs lacked integrin-aL (CD11a) and integrin-b2 (CD18) as well as integrin-αM (CD11b) and integrin-b2 (CD18), which heterodimerize to form lymphocyte function-associated antigen-1 (LFA-1) and macrophage 1 antigen (Mac-1), respectively. Although Ad-MSCs and UC-MSCs expressed integrin-a4 (CD49d) and integrin-b2 (CD29), the subunits that comprise very late antigen (VLA)-4 BM-MSCs did not express integrin-a4. Consequently, BM-MSCs may not be capable of adhering to VCAM-1, a critical step involved in extravasation of cells into the CNS during EAE (13). The hyaluronic acid receptor CD44 was highly expressed by all three MSC types.
Cell Surface Adhesion Molecules Expressed by MSCs

Cell surface expression of adhesion molecules by MSCs as assessed by flow cytometry. –, not detected; +, low (5–10%); ++, high (>80%). CD, cluster of differentiation; PSGL-1, P-selectin glycoprotein ligand-1; CAMs, cell adhesion molecules; ALCAM, activated-leukocyte cell adhesion molecule; CEACAM-1, carcinoembryonic antigen-related cell adhesion molecule-1; ICAM-1, intracellular adhesion molecule-1; MCAM, melanoma-cell adhesion molecule; NCAM, neural cell adhesion molecule; PECAM-1, platelet/endothelial cell adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1.
We next examined chemokine receptor mRNA expression by MSCs. All three MSC types expressed chemokine (C-C motif) receptor 6 (CCR6), CCR7, CCR11, chemokine (C-X-C motif) receptor 3 (CXCR3), CXCR6, and CXCR7 (Table 4). Ad-MSCs and UC-MSCs also expressed CCR9 and CCR10, while only Ad-MSCs expressed CCR1. CXCR4, which plays an important role in migration of cells to injured sites and has been shown to be expressed by MSCs (17,36), was in this case not detected. Previous studies have established that proinflammatory cytokines present during inflammation can modulate the expression of homing molecules (17,34,60). IFN-γ, the signature cytokine of Th1 cells, is upregulated during the acute phase of EAE (57). Thus, we also compared the expression of chemokine receptor mRNA by all three MSC types under standard culture conditions and following stimulation with IFN-γ for 72 h. No qualitative changes in chemokine receptor expression were observed following stimulation of BM-MSCs or Ad-MSCs with IFN-γ, while expression of CCR6 mRNA by UC-MSCs appeared to increase (Fig. 7). Given that long-term cell culture has been shown to decrease chemokine receptor expression and responsiveness to chemoattractants (36), we also analyzed mRNA from MSCs at passage 3 but found no difference in chemokine receptor expression profiles compared to passage 5 cells (data not shown).
Chemokine Receptor mRNA Expression by MSCs
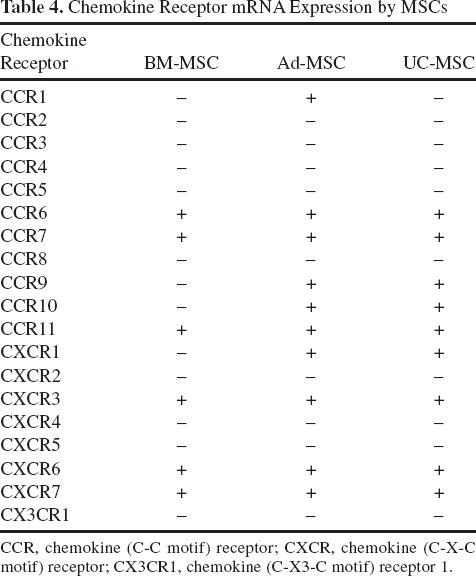
CCR, chemokine (C-C motif) receptor; CXCR, chemokine (C-X-C motif) receptor; CX3CR1, chemokine (C-X3-C motif) receptor 1.

IFN-γ has little effect on chemokine receptor mRNA expression by UC-MSCs. Passage 5 UC-MSCs were cultured in the presence or absence of 10 ng/ml IFN-γ for 72 h, and mRNA was extracted in order to detect transcripts for specific chemokine receptors. Peripheral blood mononuclear cells were used as a positive control. +/–, presence or absence of IFN-γ or reverse transcriptase (RT) in preparation of cDNA. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. See Table 4 for chemokine receptor abbreviations.
The Fate of Intravenously Transplanted MSCs
To compare the in vivo trafficking potential of BM-MSCs, Ad-MSCs, and UC-MSCs, we utilized a noninvasive bioluminescent imaging technique to examine their biodistribution in NOD/Lt mice with relapsing– remitting EAE. MSCs were transduced with a bicistronic lentiviral vector encoding firefly luciferase (fluc) and eGFP and then transplanted IV into EAE mice during the first clinical relapse (day 27 post-disease induction). Bioluminescence fluc signals were predominantly detected in the lungs of EAE mice, indicating that all three MSC types had accumulated within the pulmonary capillaries. This fluc signal diminished rapidly during the following 48 h and was undetectable 14 days post-cell injection (Fig. 8A, data not shown). No fluc signal was detected in the CNS or peripheral lymphoid tissues at any of the time points analyzed.

Ad-MSCs are capable of trafficking to the CNS in EAE mice. MSCs were transduced with a lentiviral vector encoding firefly luciferase and eGFP. (A) MSCs were transplanted IV in NOD/Lt EAE mice on day 27 postimmunization and the bioluminescence fluc signal was examined. Representative images of the distribution of MSCs 48 h after injection are shown. (B) Ad-MSCs and BM-MSCs were transplanted into C57Bl/6 EAE mice on day 7 postimmunization. DNA extracted from lymph nodes (LN), spleen (Sp), brain (Br), cerebellum (Cb), and spinal cord (SC) on day 8 and day 14 postimmunization was used as a template in PCR reactions using enhanced green fluorescent protein (eGFP) or GAPDH-specific primers. DNA from cFMS-GFP mice was used as a positive control (+ve) and DNA from uninjected mice was used as a negative control (N). Representative images of one mouse for each time point are shown.
As bioluminescence imaging may not have been sensitive enough to detect low numbers of trafficking MSCs, we next used a more sensitive technique based on the detection of eGFP+ cells by PCR to determine the tissue distribution of IV transplanted MSCs. Given that BM-MSCs lacked expression of integrin-a4, we compared the trafficking of these cells to Ad-MSCs, which were found to express high levels of this important homing molecule. Transduced BM-MSCs and Ad-MSCs were injected IV into C57Bl/6 mice on day 7 post-disease induction, and the spleen, lymph nodes, brain, cerebellum, and spinal cord were examined by PCR for the presence of the eGFP sequence on days 8 and 14 post-disease induction. eGFP+ BM-MSCs were detected in the lymph nodes of EAE mice at both time points, confirming that these cells are capable of homing to sites of inflammation in the periphery (Fig. 8B). Significantly, eGFP+ Ad-MSCs were detected in the spinal cord 24 h after transplantation as well as in the brain and lymph nodes of EAE mice 7 days after transplantation. Thus, under neuroinflammatory conditions, the broader repertoire of homing molecules expressed by Ad-MSCs appears to correlate with an enhanced homing capacity in vivo.
Discussion
Since their original identification within the BM compartment, MSCs have been identified and characterized from a wide variety of tissue sources (18). In comparison to BM aspirates, which are attained by an invasive isolation procedure (68), higher cell yields can be obtained using safer methods from other sources, such as liposuctioned adipose tissue or discarded umbilical cords. Given the abundance of these cellular resources, recent attention has been directed towards evaluating the application of alternative MSC types in a clinical setting for the treatment of inflammatory diseases. In the current study, human MSCs isolated from three different tissue sources were characterized and their therapeutic efficacy evaluated using an EAE model of chronic progressive disease induced by rMOG. Our results show a disparity between the in vitro immunomodulatory potential of MSCs and their clinical effect, highlighting that other functional properties, such as the efficiency for homing to sites of inflammation, may be critically important to the overall efficacy of MSC transplantation.
Comparative MSC studies have revealed differences in the proliferative capacity, differentiation potential, cell surface phenotype, and immunomodulatory properties of tissue-specific MSCs (37,40,55). Evidence presented in the current study clearly demonstrated that BM-MSCs exerted more potent immunomodulatory effects in vitro compared to Ad-MSCs and UC-MSCs, further confirming these reports. Only BM-MSCs demonstrated the capacity to modulate T-cell responses through soluble factors present in conditioned medium. Two recent reports have highlighted the importance of soluble factors released by MSCs in mediating clinical recovery. One study showed that mouse BM-MSCs ameliorated EAE through the paracrine secretion of chemokine (C-C motif) ligand 2 (CCL2), which, upon matrix metalloproteinase proteolytic processing into an antagonistic derivative, could inhibit the function of Th17 cells (62). The other identified hepatocyte growth factor as the major constituent of BM-MSC conditioned medium that stimulated remyelination and reduced functional deficits when administered to EAE mice at the peak of disease (2).
Our results also revealed that production of the cytokines TNF-α, IL-6, and IL-10 was significantly upregulated when 2D2 splenocytes were cocultured with BM-MSCs or BM-MSC conditioned medium. This profile was not observed with Ad-MSCs or UC-MSCs, suggesting that soluble factors secreted by BM-MSCs may account for their potent in vitro immunomodulatory effects. We did not identify the mechanism(s) by which BM-MSCs induced this cytokine secretion profile, although, given the pleiotropic activity of many cytokines, numerous pathways may be involved. TNF-α, for example, is often considered to be a proinflammatory cytokine but can, paradoxically, also exert anti-inflammatory effects (48). The increase in production of IL-6 and IL-10, which both signal through signal transducer and activator of transcription-3 (45), suggests that BM-MSCs may be modulating Treg and Th17 differentiation pathways. Alternatively, the suppressive effect exerted by MSCs on T-cells may occur indirectly via the modulation of antigen-presenting cell function and induction of IL-10 production, as has been shown previously (33).
On the basis of our in vitro characterization studies, it was predicted that BM-MSCs would have a greater therapeutic efficacy when transplanted into an autoimmune-mediated disease setting. However, our results showed no significant difference in the clinical recovery or suppression of peripheral T-cell responses in EAE mice treated with BM-MSCs, Ad-MSCs, or UC-MSCs. In fact, only transplanted Ad-MSCs generated a significant reduction in disease severity and CNS pathology. The lack of correlation between the ability of BM-MSCs to suppress immune responses in vitro and the ability to induce clinical improvements in vivo has been reported in other disease settings, including graft versus host disease (69), collagen-induced arthritis (66), and experimental autoimmune neuritis (65). To understand why BM-MSCs did not potently suppress disease signs in EAE, we explored the expression of homing molecules and receptors involved in chemotaxis and migration. Overall, we found that Ad-MSCs and UC-MSCs expressed a broader repertoire of homing molecules compared to BM-MSCs. All three MSCs lacked expression of P-selectin glycoprotein ligand-1, which interacts with E- or P-selectin on inflamed endothelial cells and mediates the initial tethering and rolling of circulating leukocytes (64). However, CD44, which may be an alternative mechanism utilized by MSCs for these primary migratory events (10) through binding to hyaluronic acid (19), was expressed by all three MSCs types. Integrins mediate firm adhesion of circulating cells to endothelial cells via binding membrane-bound molecules such as VCAM-1 and ICAM-1, which are upregulated during inflammation (13). Binding of VLA-4 (integrin-a4b1) on migrating T-cells to VCAM-1 on brain endothelium is important for T-cell extravasation across the blood–brain barrier, as indicated by the efficacy of the blocking antibody natalizumab (12). In concert with its effects on T-cells, Pluchino et al. (59) and Constantin et al. (14) provided evidence for the importance of VLA-4 in transplanted neural precursor cells and MSCs, respectively, to migrate across the blood– brain barrier in EAE. We detected integrin-b1 expression on all three MSCs, but only Ad-MSCs and UC-MSCs expressed integrin-α4. This finding is consistent with a previous report demonstrating a lack of surface integrin-α4 expression on BM-MSCs (20), rendering these cells incapable of functionally binding to VCAM-1. Although the molecular trafficking mechanisms utilized by MSCs to home to sites of inflammation in EAE are not fully resolved, the lack of integrin-a4 expression by the BM-MSCs used in the present study may limit the migratory potential and therapeutic efficacy of these cells, regardless of their potent immunomodulatory properties. Indeed, results presented herein examining the fate of MSCs in rMOG-EAE revealed that Ad-MSCs were capable of trafficking to the spinal cord and brain while BM-MSCs were not. Likewise, a recent report demonstrated that engraftment of MSCs and neural precursor cells in a rat model of traumatic brain injury was highly dependent on the interaction between integrin-α4 and VCAM-1 (49). Expression of integrin-α4 is particularly relevant in the current disease setting, given that all three MSC types did not express other integrin heterodimers important for leukocyte trafficking to inflammatory sites in EAE, such as LFA-1 (integrin-αLβ2) and Mac-1 (integrin-αMβ2).
Numerous chemokines and their receptors have been implicated in leukocyte trafficking to peripheral lymphoid tissues and the CNS in EAE, including CCR1, CCR2, CCR5, CCR6, CCR7, CXCR3, and CXCR4 (35,64). Our analysis of chemokine receptors showed that all three MSCs expressed mRNA for CCR6, CCR7, CCR11, and CXCR3, while only Ad-MSCs expressed CCR1. Other studies have also investigated chemokine receptor expression by MSCs, which, together with our data, suggest that expression profiles can vary between tissue sources and donors. For instance, we did not detect expression of CXCR4 mRNA by any of the three MSC types. This is consistent with a recent comparison of BM-MSCs and UC-MSCs (4), while previous reports have demonstrated expression of CXCR4 on MSCs (10,36). In addition to donor variability, culture conditions can influence the expression of homing molecules and migratory capacity of MSCs. Honczarenko et al. (36) found a loss of chemokine receptor expression and chemotactic responses of MSCs due to long-term culture as well as loss of ICAM-1, ICAM-2, VCAM-1 expression. Similarly, Tondreau et al. (70) reported a loss of VCAM-1 and metalloproteinase expression by MSCs between passages 3 and 5. In contrast to these findings, we observed no changes in chemokine receptor expression between passages 3 and 5 MSCs. Exposure to cytokines and growth factors has been shown to upregulate homing molecules and stimulate cellular migration. For instance, platelet-derived growth factor can increase surface expression of CD44 on the rat MSC line Ap8c3 (76). In the present study, we found that stimulation with IFN-γ also had very little effect on chemokine receptor mRNA expression. Although Hemeda et al. (34) reported a similar finding following IFN-γ stimulation, exposure to TNF-α was shown to regulate MSC migration. Further investigations into the regulatory effects of culture conditions and cytokines on MSC migration using multiple donors are required.
Our bioluminescence tracking studies confirmed previous reports demonstrating that the majority of MSCs are arrested in the pulmonary vascular beds following IV administration (27). Preclinical animal studies in EAE and other disease models have nevertheless indicated that clinical improvement does not require high levels of MSC engraftment but may often be associated with a “touch-and-go” effect (72). This may involve direct interactions with immune cells and/or paracrine secretion of soluble immunomodulatory and trophic factors early after infusion, but with long-lasting effects. The results from our transplantation studies revealed that multiple injections of MSCs before disease onset led to an improved clinical outcome in EAE. The reduced proliferative response of splenic T-cells following MSC treatment and the enhanced therapeutic effect of high-dose IP Ad-MSC transplantation in NOD/Lt mice supports a peripheral immunosuppressive mechanism of action. Ad-MSCs may have also exerted anti-inflammatory effects within the CNS of EAE mice, leading to a significant decrease in disease severity compared to PBS-injected controls. Immune modulation is, however, not the only mechanism by which MSCs can induce clinical recovery in EAE. For instance, paracrine secretion of growth factors such as vascular endothelial growth factor, insulin-like growth factor, hepatocyte growth factor, and fibroblast growth factor by MSCs may contribute to antiapoptotic, antioxidant, and antiscarring effects (46).
While the increased efficacy of early MSC treatment we observed is consistent with that shown by Zappia et al. (75), these results differ from previous reports (14,62), in that MSC treatment from the onset of clinical signs had no impact on the disease course. This could be due to several reasons, such as the use of rMOG to induce EAE rather than the encephalitogenic MOG35–55 peptide commonly used in other studies. Although disease is initiated by CD4+ T-cells in both models, antigen-activated B-cells produced in rMOG-EAE contribute to disease pathogenesis by both humoral and cell-mediated mechanisms. B-cells act as antigen presenting cells to promote the differentiation of MOG-specific Th1 and Th17 T-cells (73) and are a major source of inflammatory cytokines (5). Moreover, antibodies that recognize conformational but not linear MOG epitopes can mediate demyelination (50). MSCs have been reported to inhibit B-cell function, suppressing their proliferation, differentiation, chemotaxis, and immunoglobulin production (15). In the present study, we show that while the nonspecific and rMOG-specific proliferative response of splenic T-cells is significantly reduced, MSC transplantation appears to have no impact on serum rMOG-specific antibody levels in this model. Interestingly, in addition to their pathogenic role, B-cells may also act as regulators of CNS autoimmunity through producing IL-10 and influencing forkhead box P3-positive (FoxP3+) Treg homoeostasis (25). Crosstalk between MSCs and inflammatory signals received from the tissue microenvironment plays an important role in directing the nature of the MSC response. For instance, in models of Th1/Th17-driven inflammation such as EAE, MSCs can induce clinical recovery through suppressing Th1/Th17 responses and promoting a Th2 type cytokine shift (3,14), whereas their efficacy in Th2-mediated allergic airway inflammation results from the promotion of a Th1 phenotype (30). Given the recent concept that B-cells may have a dual function in EAE, their interaction with MSCs, and how this bidirectional crosstalk may influence pathogenic versus protective B-cell responses warrants closer examination.
The MSC treatment paradigms we investigated collectively suggest that timing as well as cell dose and delivery route influence MSC efficacy in rMOG-EAE. MSC transplantation at the onset of disease had no impact on chronic progressive EAE, while a single dose before disease onset was also not sufficient to alter the clinical course. Using the relapsing–remitting model, we found that IP delivery of a high Ad-MSC dose prevented disease-related mortality, whereas a low IV cell dose did not. Preclinical studies in graft versus host disease have also shown that the kinetics of MSC delivery is critical to their therapeutic efficacy (42). “Licensing” of MSCs by proinflammatory mediators, which modulate the MSC secretome and increase production of inhibitory and trophic factors, may be necessary in order to achieve beneficial outcomes (42) and will be affected by the timing and route of administration. Thus, we hypothesize that the low number of IV transplanted MSCs that were capable of reaching inflammatory sites were not sufficient to attenuate rMOG-EAE severity when cells were transplanted later in the disease course or at a single dose. Moreover, although MSCs have shown efficacy in numerous disease models within the context of a xenogeneic transplantation setting, we cannot rule out the possibility that clearance of transplanted MSCs by the host immune system was a further contributing factor that limited their therapeutic effect in rMOG-EAE mice.
In summary, we compared three tissue-specific human MSCs and demonstrated that BM-MSCs exerted more potent immunomodulatory effects in vitro compared to Ad-MSCs and UC-MSCs. Unexpectedly, BM-MSCs did not impact the disease course; however, transplantation of Ad-MSCs ameliorated clinical signs in two animal models of EAE. Significantly, the timing and dose of cell transplantation were found to be the most critical factors that led to improved clinical and pathological outcomes. The disparity between our in vitro and in vivo findings may reflect differences in the homing potential of the MSCs, as suggested by differential expression of integrin-a4 and the inability of BM-MSCs to traffic to the CNS in the absence of this adhesion molecule. While the immunomodulatory properties of MSCs play a critical role in mediating the amelioration of disease in EAE, the efficiency of MSC homing is also likely to be a key determinant of therapeutic efficacy (38). Modification of MSCs represents one means by which directed homing can be improved. For instance, forced expression of integrin-a4, which was absent on the BM-MSCs used in this study, improved homing to bone in immunocompetent mice (44). Understanding the fate of MSCs in vivo and the molecular mechanisms controlling their migration will therefore be critical for improving the therapeutic efficacy of systemic MSC transplantation for the treatment of MS.
Footnotes
Acknowledgments
We thank BD Biosciences for their generous provision of the Human Cell Surface Marker Screening Panel test kit. This work was supported by grants from the Baker Foundation, the National Health and Medical Research Council of Australia, the Diana Asmar Fund, the Eva and Les Erdi AUSiMED Fellowship in Neurological Diseases, Multiple Sclerosis Research Australia, and Cure Multiple Sclerosis Fellowship-Neurological Diseases. N.P. was supported by a Trish MS Research Foundation Postgraduate Research Scholarship and a Monash University Postgraduate Publication Award. The authors declare no conflicts of interest.