
Abstract
Transplantation therapies aimed at repairing neurodegenerative and neuropathological conditions of the central nervous system (CNS) have utilized and tested a variety of cell candidates, each with its own unique set of advantages and disadvantages. The use and popularity of each cell type is guided by a number of factors including the nature of the experimental model, neuroprotection capacity, the ability to promote plasticity and guided axonal growth, and the cells' myelination capability. The promise of stem cells, with their reported ability to give rise to neuronal lineages to replace lost endogenous cells and myelin, integrate into host tissue, restore functional connectivity, and provide trophic support to enhance and direct intrinsic regenerative ability, has been seen as a most encouraging step forward. The advent of the induced pluripotent stem cell (iPSC), which represents the ability to “reprogram” somatic cells into a pluripotent state, hails the arrival of a new cell transplantation candidate for potential clinical application in therapies designed to promote repair and/or regeneration of the CNS. Since the initial development of iPSC technology, these cells have been extensively characterized in vitro and in a number of pathological conditions and were originally reported to be equivalent to embryonic stem cells (ESCs). This review highlights emerging evidence that suggests iPSCs are not necessarily indistinguishable from ESCs and may occupy a different “state” of pluripotency with differences in gene expression, methylation patterns, and genomic aberrations, which may reflect incomplete reprogramming and may therefore impact on the regenerative potential of these donor cells in therapies. It also highlights the limitations of current technologies used to generate these cells. Moreover, we provide a systematic review of the state of play with regard to the use of iPSCs in the treatment of neurodegenerative and neuropathological conditions. The importance of balancing the promise of this transplantation candidate in the light of these emerging properties is crucial as the potential application in the clinical setting approaches. The first of three sections in this review discusses (A) the pathophysiology of spinal cord injury (SCI) and how stem cell therapies can positively alter the pathology in experimental SCI. Part B summarizes (i) the available technologies to deliver transgenes to generate iPSCs and (ii) recent data comparing iPSCs to ESCs in terms of characteristics and molecular composition. Lastly, in (C) we evaluate iPSC-based therapies as a candidate to treat SCI on the basis of their neurite induction capability compared to embryonic stem cells and provide a summary of available in vivo data of iPSCs used in SCI and other disease models.
Pathophysiology of Spinal Cord Injury and Stem Cell Therapies
Pathophysiology of Spinal Cord Injury (SCI)
Most human spinal cord injuries (SCI) are contusions resulting in dislocation of vertebral columns after an initial traumatic event. These injuries are diffuse and result in central cavitation surrounded by spared white matter. SCI follows a biphasic pattern referred to as “primary” and “secondary” phases. SCI involves extensive interaction of various immune cells, resident CNS cells, noncellular components (such as adhesion molecules, cytokines, and chemokines) to mount inflammatory, immune, and scar tissue responses resulting in extensive tissue destruction, cyst formation, scar tissue formation, Wallerian degeneration, and Schwannosis (45). Most details on the pathophysiology of SCI have been obtained from experimental SCI in animal models as well as stereotyped responses from traumatic brain injury. Despite extensive data detailing the events of experimental SCI [for reviews, see refs. (96,108, 168,248,249,297,306,318,324,333)] the exact course of events in human SCI is less clear. There seem to be basic similarities, but there is evidence that fundamental differences may exist, such as the extent and importance of the astrocytic response and demyelination (270).
The primary phase consists of the mechanical compression of tissues and nerves, which causes membranes to rupture (326). The extent of axonal death rises exponentially with the initial force of impact (109) and the sparing of fibers is reflected in locomotor deficits (168,331). Complete spinal cord transection is rare, and most incomplete or contusion human injuries spare a rim of mostly demyelinated peripheral axons (27,43,382). Generalized immediate spinal cord swelling is usually the first visible sign of injury, disrupting normal blood flow and causing hypoxia, usually accompanied by hemorrhage (41,46,96,193,303). Mechanical factors are also in part responsible for damage to the blood–brain barrier (BBB), which increases permeability, facilitates peripheral cell invasion into the lesion and contributes to swelling and inflammatory events.
The primary phase of events responsible for the immediate wave of cell death is followed by a secondary phase, which results in widespread and prolonged tissue destruction mediated by excitotoxic, oxidative, inflammatory, and immune events. These are interlinked events, poorly defined especially in humans, and animal models seem to have species and strain specific differences as well as differences depending on the type of injury (47,96,111, 177,298,342). Together, these factors are typically far more damaging than that of the primary phase and can affect surrounding cells up to several millimeters from the lesion epicentre in rodents and several centimeters in humans.
Excitotoxicity and Oxidative Damage
Ionic imbalances of key ions (such as sodium, potassium, and calcium) and subsequent excessive depolarization are a product of ischemia, hypoxia, and hemorrhage (288,314). This contributes to excitotoxicity (excessive stimulation by neurotransmitters such as glutamate), formation of reactive oxidative species (ROS), and lipid peroxidation (55,175,219,365). These events cause loss of osmotic balance, disruption of cell membranes, altered mitochondrial and metabolic activity, leading to widespread cell death and axonal degeneration (377).
Inflammatory and Immune Responses
The damaged BBB allows influx of leukocytes, triggering an inflammatory response (170). Microglia and macrophages are elevated almost immediately post-SCI, peak at about 8 h and remain elevated for weeks postinjury (96,205). These cells slowly clear debris by phagocytosis, participate in scar tissue formation, cause neurotoxic “bystander damage,” and secrete various neuroprotective factors. Neutrophil infiltration peaks at about 24 h post-SCI (96,111), returns to normal levels within the first week postinjury, and causes neuronal and glial toxicity by release of cytokines, ROS, and neurotoxic enzymes. Lymphocytes are elevated by 3–4 days post-SCI and peak by 7 days. Cytokines (including tumor necrosis factor-α, interleukins-1β and -6) are released from inflammatory cells (as well as neuronal and glial cells), peak within 1–6 h and decline by 1 week postinjury (96). These cytokines exacerbate inflammation and BBB permeability, promote apoptosis and necrosis of neurons and oligodendrocytes, reduce axonal growth, and may impair locomotor recovery (194).
Cavity/Cyst Formation
Contusion injuries are characterized by formation of cystic structures, which arise from the clearance of debris by macrophages and progressive loss of neural tissue (19,111,270). A spared rim of white matter usually persists, but cysts represent a physical barrier that inhibits neurite regeneration. The cysts are delineated by the formation of scar tissue, which also eventually becomes inhibitory.
Scar Tissue Formation
Scar tissue is now widely accepted to be both a physical as well as a chemical barrier for axonal growth and regeneration, at least in experimental SCI (108,306,320). Its composition as well as spatiotemporal distribution is extremely dynamic and malleable within the first few weeks postinjury (186,364) at which point the composition stabilizes and does not change (“chronic injury”). Scar tissue is mainly composed of macrophages, microglial cells, pericytes, oligodendrocytes precursor cells (OPCs), oligodendrocytes, meningeal cells, and macrophages, each of which express/secrete several growth inhibitory molecules. The key molecular components include extracellular matrix components (especially collagens), sulfate proteoglycans (including chondroitin sulfate, heparin sulfate, and keratin sulfate), tenascins, semaphorins, and ephrins [for reviews, see refs. (13,81,108,110,123,186)]. Inhibitory molecules within the glial scar are important contributors to abortive neuronal regeneration and may cause collapse of axonal growth cones (9,86,87,165,250,290); thus, the reduction of scar tissue is a commonly used indicator of morphological improvements in experimental SCI. However, it is equally well accepted that cells participating in scar tissue formation, especially astrocytes, also have a neuroprotective role by secreting growth promoting and/or neuroprotective factors (36,306,333,388). Overall, axonal growth after SCI depends on (i) molecular and spatiotemporal factors that balance inhibitory and growth promoting influences (165), (ii) the subtype of axon that is involved (e.g., serotonergic fibers rarely grow into scar tissue) (88,139,140,155,309), and (iii) the intrinsic growth state of the mature neuron.
Wallerian Degeneration and Demyelination
Degeneration of the axon and myelin sheath distal to the injury after axonal disconnection with the cell body is followed by slow removal of neural tissue debris by macrophages and microglia cells (4,20,102,270,304,418). This is a protracted process in humans, beginning 3–6 days postinjury and becoming more extensive after 12–15 days postinjury (48,102). After 1 year postinjury, there is almost a complete loss of axons in affected tracts (48). Oligodendrocytes undergo apoptosis following SCI in both experimental and human SCI (83,102,112,247). Rapid loss of the oligodendrocyte population leads to demyelination (in experimental animal models within 1.5–4 h postinjury) and loss of remyelination (48,369). The resulting extensive myelin debris renders the injury site an effective inhibitor to axonal regrowth. Furthermore, demyelination directly contributes to dysfunction of axons and action potential transmission (264). Yet, the extent and importance of demyelination in human SCI is unclear. Primary demyelination, which is the destruction of myelin while still obtaining intact axons, seems to be uncommon in humans (47,168,270), but demyelination associated with Wallerian degeneration is more common (4). Several authors have reported a gradual loss of myelin, but not to the same degree in all patients (48,125).
Current Stem Cell Therapies in Spinal Cord Injury
Cellular transplantation approaches are aimed at targeting the pathophysiology of SCI, in particular to replace lost endogenous neuronal and/or glial cells, provide a more favorable growth environment to mask or neutralize inhibitory molecules, and enhance and direct any intrinsic regenerative ability that neurons may possess (17,229,233,317) (Fig. 1). Traditional cell transplants commonly used in experimental SCI are neural stem/progenitor cells, glial precursors, olfactory ensheathing glial cells, mesenchymal stem cells, and Schwann cells, each of which can produce some morphological or behavioral benefits (for review, see ref. 367).
The delayed loss of myelinating oligodendrocytes provides another problem that can realistically benefit from cellular transplants. Replacing lost endogenous oligodendrocytes with donor oligodendrocyte precursor cells (OPCs) or a mixed transplant of neural stem/precursor cells can limit damage and potentially restore function by replacing myelinating oligodendrocytes. Specifically, studies have found transplantation of OPCs results in differentiation to mature oligodendrocytes, which can increase myelination up to 50%, and correlates with improvements in locomotor function (33,52,80,106,157,176,254,328). However, obtaining large quantities of purified human OPCs or neural stem/progenitor cells (which can give rise to OPCs) is problematic. Embryonic stem cell (ESC) lines that can be differentiated towards neural or glial lineages are a useful option because they can yield very large quantities of donor OPCs, which cannot be easily obtained from adult stem cell sources (120). Yet, embryonic cell lines are not genetically identical to the patient and as such donors must be carefully immunologically matched to recipients, along with appropriate immunosuppression therapies that are known to have adverse long term effects (224). Furthermore, the use of ESCs is associated with ethical controversy (137,159,310,313). Somatic cell nuclear transfer (SCNT) and cell fusions (144,158,251,300,308,401) are alternative sources of pluripotent stem cells, but these are technically challenging, inefficient, costly, rely on donor oocytes (and as such also introduce ethical issues), and although it can produce cells with cloned nuclear DNA, the mitochondrial DNA remains of maternal origin which may be sufficient to elicit immune rejection (156).

Stem cell transplants in SCI. The aim of stem cell transplants is to attenuate the pathology of SCI by replacing lost endogenous cells and functions, thereby influencing the local milieu and microenvironment to improve neuronal plasticity and promote functional recovery.
The discovery that somatic adult cells could be dedifferentiated (or “reprogrammed”) back into an ESC-like state (termed induced pluripotent stem cells or “iPSCs”) has been an important milestone in clinical regenerative research (362,410). iPSCs offer potential solutions to the various problems described above by providing an almost unlimited source of OPCs or other desirable cells from adult tissue, human iPSCs can be obtained by simple noninvasive procedures and delivered as an autograft, which is likely to reduce host immune responses (98,291,356). Furthermore, iPSCs have now been shown to reliably differentiate into definite neural lineages (see “Can iPSCs Form Functional Neurons? Neurite Induction Compared to ESCs”). This makes iPSC technology a feasible new alternative for cell-based therapy for many CNS conditions including SCI. iPSC technology offers solutions to the ethical issues invariably associated with the use of human ESCs (although iPSCs are themselves not free of ethical controversies) (44,84,231,336), as well as offering autologous transplant possibilities that may attenuate rejection problems.
Generation and Characteristics of Induced Pluripotent Stem Cells
What Are Induced Pluripotent Stem Cells? An Introduction to the Technology
iPSCs are generated by introducing “reprogramming” factors involved in maintenance of pluripotency and self-renewal (39,67,163,182,223,360,362). Originally, four transcription factors [octamer-binding transcription factor 3/4 (oct3/4), sex-determining region Y box 2 (sox2), Krüppel-like factor (klf-4), and c-myc] were used to establish the first iPSCs from mouse embryonic fibroblasts (MEFs) (362). These initial attempts yielded partially reprogrammed iPSCs (compared to the current standard; see “Criteria of Bona Fide iPSCs: Measuring Pluripotency Experimentally”) as they could not give rise to live chimeric mice and thus did not demonstrate authentic pluripotency. The field quickly advanced and soon improved protocols with variations of reprogramming factors (importantly, the oncogene c-myc was shown to be dispensable) (260,385), selection processes of iPSC clones [more appropriate selection markers such as nanog and oct3/4 (232,279,386) instead of F-box protein 15 (fbx15) (362) or no selection at all (26)] began to yield high-quality fully pluripotent iPSCs with silenced transgenes and the ability to form live organisms by both chimera formation and tetraploid complementation, the most stringent experimental assessments of pluripotency (see “Criteria of Bona Fide iPSCs: Measuring Pluripotency Experimentally”). This has now been achieved with a wide range of species including rats (208,213), humans (361,410), pigs (103,104,395), sheep (218), horse (259), nonhuman primates (59,89,422), and endangered wildlife species (24).
The underlying molecular mechanisms of pluripotency and reprogramming are complex interactions between transcription factors, epigenetic regulators, signaling pathways, and microRNA interactions (39,64,65,158,223,228,253, 295,360,394,400), which are not detailed in this review. The reprogramming factors act to induce pluripotency synergistically, requiring a delicate balance of factors for a sustained period of time (~10–12 days) to reprogram the cells (54,286,344,362). It is unclear why only small proportions of cells exposed to reprogramming factors become fully programmed (typically 0.0001–2.0%, depending on the delivery method used—which albeit low, is still more efficient than other methods to create pluripotent stem cells). Several models have been proposed to explain these low efficiencies, including a stochastic model (correct levels of expression of the factors occur by chance in small amounts and potent resistant forces such as epigenetic landscape, tumor suppressors, or senescence inductors oppose pluripotency) and an elite model (stating as yet unidentifiable cells or stem cells present within the culture at low frequencies are able to be reprogrammed or overcome barriers to pluripotency) (130,228,295,399), although given enough time all cells eventually become amenable to reprogramming (for review, see ref. 295). Nevertheless, upon correct exposure of reprogramming factors to cells, the sequential activation of endogenous pluripotent markers follows, marking the onset of pluripotency; alkaline phosphatase, stage-specific embryonic antigen (SSEA)-1, followed by oct3/4, and nanog.
The transcriptional network governing pluripotency appears to be conserved across species, because human or mouse transgenes can be used to reprogram other species, and is not restricted by the germ layer of origin of the parental donor cell. The resultant iPSC cells share the same phenotypic markers and characteristics as ESCs—they are pluripotent, self-renewing, and rapidly proliferate in culture—features that links these cells to tumorigenicity (see “Tumorigenicity of iPSCs”). Originally, iPSCs were deemed almost indistinguishable from ESCs in terms of genetic and epigenetic composition, but with recent reports it is now increasingly apparent that iPSCs are neither identical to ESCs nor identical to other iPSC lines and may still bear a “memory” of their parental donor cell (see “Molecular Analysis of iPSCs”). The full implications of these differences are unknown at this time.
Generation of iPSCs: Methods of Transgene Delivery
Originally iPSCs were produced by delivering sox-2, oct3/4, klf-4, and c-myc transcription factors by retroviral transduction, resulting in permanent genomic integration (362). Since then, much research has focused on improving the reprogramming method (232,252,260, 279,361,385,386,410). From a clinical perspective, the ideal method would reliably generate safe iPSC clones at a high rate (i.e., efficient reprogramming) without inclusion of oncogenes or requiring permanent genome integration, and can be derived xeno-free, is a technically simple and cost-effective process (Table 1), and cells can be produced in a time frame optimal for therapeutic use.
Arsenal of Current Technologies Available to Produce iPSCs

References are not an exhaustive list especially for viral-integrating methods [for reviews see refs. (65,128)]. Efficiency of reprogramming can be enhanced with: (i) incorporation of additional reprogramming factors (e.g. lin28, nanog, glis1) (421), (ii) downregulation/knockout of tumor suppressor related genes (e.g. p53, p16, p15, p19, INK4) (16,147,174,206,240,280,374,421), (iii) modification of cell-signalling (e.g. Wnt Pathway) (242), (iv) treatment with chemicals and small molecules (e.g. Valcroic acid, kenpaullone, thiazovivin, SB431542, PD0325901, vitamin C, Ps48, lithium, butyrate) (103, 151,211,215,227,232,235,330,378,428), (v) addition of anti-apoptotic agents (e.g. SV40) (289), (vi) silencing of apoptotic inducers (e.g. Puma) (195), (vii) selection process for fully pluripotent stem cells [e.g. drug selection for nanog, oct-4 (232,279,386) or reporter system with miRNAs (94)] and (viii) hypoxic conditions (374,408). Combining several of these “enhancers” can reduce the time needed for reprogramming (332). Note: rate of iPSC formation (not efficiency) can be increased by cell division, as this results in increased cumulative probability for the stochastic events to occur earlier in time (130). ESC, embryonic stem cell; iPSC, induced pluripotent stem cell; miRNA, micro RNA; OSKM, oct4, sox2, klf-4, c-myc.
Choice of Transcription Factors
It is possible to vary the number and combinations of the transcription factors used, which may be critical for clinical applications. At least one transcription factor (typically oct4) is usually required, although several studies have used synthetic messenger RNA (mRNA) (381), proteins (76,179,424), or more recently microRNAs (miRNA) that circumvent the use of these transcription factors entirely (8,214). Omission of the oncogene c-myc is desirable to reduce tumor concerns but has been linked to significantly lower reprogramming efficiency (but notably promoting growth of fewer non-iPSC colonies or “partially reprogrammed” iPSCs) (260) and may affect development potential (10). Generally, methods employing fewer transcription factors are less efficient compared to the traditional four factors, at least for fibroblasts. C-myc can be substituted with n-myc or l-myc (26,261,280). l-myc has been found to specifically enhance the number of fully reprogrammed colonies (more so in humans than in mice), whereas c-myc enhances the numbers of both fully reprogrammed iPSCs as well as partially reprogrammed clones. As such, the overall effect of l-myc is to increase numbers of iPSC clones, without increasing tumorigenicity (261). Yet, l-myc is not typically used in the majority of iPSC studies. Sox-2 can be substituted by sox-3, 7, 15, 17, or 18 (260) or chemicals (153), and klf-4 can be replaced by 18 other factors but at much lower reprogramming efficiencies (230,260). Other transcription factors such as nanog, Lin28, glioma-associated oncogene similar 1 (Glis1), B lymphoma Mo-MLV insertion region 1 (Bmi1), and T Box 3 (tbx3) can also be successfully used (127,230,257,410). No direct substitution for oct3/4 has been found to date (230,360).
Permanent Genomic Integration: Retroviral and Lentiviral Delivery
Viral integration methods with retrovirus or lentivirus are the most technically simple, common, and reliable method, reprogramming cells into iPSCs at high efficiency and providing sustained levels and durations of factor expression required for successful reprogramming. Viral delivery has proved effective with a wide variety of different donor species and cell types, tissue culture platforms, and omission of oncogenes (both c-myc and klf-4). Yet safety concerns associated with permanent genomic integration and modification make this method of transgene delivery unlikely to be used in a clinical setting. A typical retroviral or lentiviral transfection will leave up to 5–20 integration sites in the genome (241,362,390). Random integration of virus may cause insertional mutagenesis and unpredictable genetic dysfunction. Residual exogenous factors may also be reactivated, linking to high rates of teratoma formation in vivo (145,279), and alterations in molecular characteristics of iPSCs, inhibition of complete iPSC maturation, and potential differences of development capacity (339,410). Changes in vector design can help reduce these risks by reducing the number of integrations and controlling the expression and activation of the transgenes. Using polycistronic vectors (single cassette carrying all the transgenes under the control of a single promoter) can reduce the number of integration sites down to one (53,61,338–340), ensure that cells will receive all transgenes at the same time to promote more efficient reprogramming, and reduce numbers of partially reprogrammed iPSCs (121,281,339,340). Alternatively, phage integrase can be used for site-specific integration (406). The temporal timing of expression/activation of the reprogramming factors can be controlled using tetracycline-inducible promoters, which can significantly enhance reprogramming efficiency (40,128,384). MicroRNA (miRNA) can also be used to generate iPSCs (8,166,212,214,293,349). Several miRNA clusters have been found to be important regulators of the cell cycle and self-renewal and are specific to pluripotent stem cells (380). Recently, several clusters have been identified to be important in iPSC reprogramming, and delivery of these clusters in addition to the reprogramming factors (oct3/4, sox-2, klf-4 with and without c-myc) was found to considerably enhance the reprogramming efficiency (166,212,214,293,349). Notably, the miR-302 cluster has been found to be sufficient to induce reprogramming alone at high efficiency, without requiring any additional reprogramming factors (8,214).
Excision Systems: Cre-Lox and Transposons
To overcome problems associated with permanent genomic insertions, excisable systems to remove the transgenes can be used while still maintaining high reprogramming efficiencies. These include the cre-lox excision system and Transposons PiggyBac system, both of which use enzymes to remove the inserted transgenes. The cre-lox system was initially used as a means to investigate iPSC quality and pluripotency after removal of the transgenes (61,167,338,339). Following removal, iPSCs remain pluripotent and may exhibit improved development potential compared to their viral integrated counterpart (339). However, residual lox-P sites following excision of transgenes with cre-recombinase may still pose a safety risk by potentially disrupting exogenous gene function. PiggyBac transposon allows the traceless excision of transgenes (167,392,411), but transposition can also result in chromosomal rearrangement around the excision site. Both excision systems may require clonal expansion of iPSC colonies to ensure only successfully excised clones are propagated.
Transient Transgene Expression Systems
Transient expression of the transgenes (categorized into DNA-based and DNA-free vectors) can overcome problems associated with genomic integrations of imperfect excision systems but are more difficult to achieve, as successful reprogramming requires sustained level of the transcription factors for 10–12 days. This may necessitate carefully timed multiple transfections, according to the half-life of the vector. Too low a concentration of transgene levels will promote incomplete reprogramming of iPSCs and promote differentiation, and too high a concentration can be toxic to target cells.
DNA vectors [e.g., adenovirus (281,345,427), plasmids/ episomal plasmids (121,199,278,280,281,409), and minicircles (162)] confer very low efficiency of reprogramming, require multiple transfections due to vector dilution with cell division, and bear the risk of genomic integration. As such, iPSC clones derived by these methods still need to be extensively characterized for lack of integration. DNA-free vectors [including Sendai virus (15,115,327), mRNA (381,398), and protein delivery (76,179,424)] bear no risk of genomic integration. Sendai virus is an RNA-based vector that can reprogram donor cells to iPSCs at medium to high efficiency. Yet Sendai virus continually replicates in the cytoplasm where it poses a safety risk and must be removed (15,327). mRNA delivery is a highly efficient, albeit labor-intensive, method (381,398). Protein delivery using purified recombinant proteins ligated to small delivery molecules delivered directly into the cells is very inefficient, is technically complex, requires multiple carefully timed reapplications, but represents a safe delivery option without viral vectors (179,424). Protein delivery using undefined combinations of unpurified combinations of protein extract, isolated from ESCs (76), is much more efficient and reportedly does not require the normal duration of reprogramming factors; a single application is sufficient for successful reprogramming.
Generation of iPSCs: General Considerations of Delivery Approaches
It is unlikely that methods involving genomic integrations will be routinely used in a clinical setting, but the low-efficiency and labor-intensive approach of most transient methods also represent an important barrier (see Fig. 2). Permanently integrated transgenes may not be problematic in a research setting, but it should be kept in mind that transgene-free iPSCs may more closely resemble ESCs (381). Regardless of approach, it is important that each new method is tested on both human and nonhuman donor cells. Murine cells are generally reprogrammed with greater ease and efficiency than human cells and require less exposure to transcription factors. Furthermore, human donor cells cannot be subjected to the most stringent pluripotency tests (tetraploid complementation and chimera formation), and each new technology must prove that it is able to establish bona fide iPSCs according to the strictest pluripotency criterion (see “Criteria of Bona Fide iPSCs: Measuring Pluripotency Experimentally”). Different donor cell types should also be tested, as the efficiency of reprogramming also depends on the donor cell type. Keratinocytes have been shown to have one of the highest efficiency to date (1,2). Progenitor cells, especially adipose progenitor cells or neuronal progenitor cells, are also easily reprogrammed (and can efficiently reprogrammed with just oct4) whereas adult fibroblasts have a much lower reprogramming rate (183,329,353). Finally, the efficiency of reprogramming also depends on the culture platform used; the majority of studies focusing on iPSC reprogramming methods use a feeder-based system, which is generally more efficient than feeder-independent methods. Feeder-free and xeno-free derivation of iPSCs are important for potential clinical applications (312,316) and may reduce variability found among different iPSC lines (79).
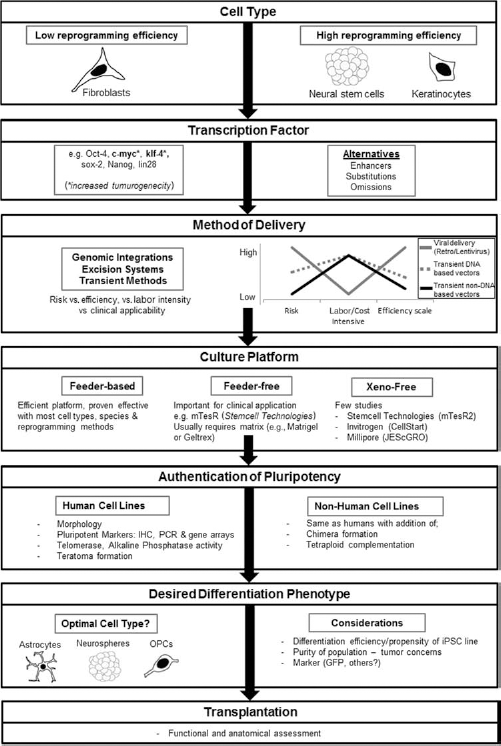
Stepwise considerations for using iPSC-derived cells for transplantations aimed at repairing SCI and other neurodegenerative and neuropathological conditions of the CNS.
Characteristics of iPSCs: A Comparison to ESCs
Criteria of Bona Fide iPSCs: Measuring Pluripotency Experimentally
Once iPSCs have been generated, pluripotency must be assessed. Pluripotent ESCs have a unique morphology, genetic, and epigenetic signature compared to somatic cells (116). As pluripotent cells, iPSCs should be similar to ESCs. Various assays are available to validate iPSCs for pluripotency—all of which should be employed consistently—at least in nonhuman iPSCs (Table 2). To satisfy these criteria is important as inconsistencies in reprogramming can result, especially in transient transgene delivery methods. These “partially reprogrammed” iPSC lines are not fully pluripotent and exist in an undefined state, expressing some pluripotent markers but not others, and have limited differentiation potential (60). Importantly, lines derived from the same parental donor cell, using the same derivation method, can show differences in functional (differentiation) potential (30,169,343). From this, it is becoming increasingly clear that the pluripotent state can be dynamic and varied, thus a common standard to assess pluripotency should be used for assessment of new cell lines and which is becoming more critical as more technologies to create iPSCs are being developed (337).
iPSC Lines Should Be Validated for Pluripotency by These Standard Criteria

Morphology and pluripotent marker expression is often used as a selection criterion to selectively propagate fully pluripotent iPSCs from surrounding nonreprogrammed cells (362,414). iPSCs express the same surface antigens as ESCs. Notably, no single marker can be reliably used to distinguish bona fide iPSCs from heterogeneous populations of partially reprogrammed cells (60). Chan et al. (60) found that markers commonly used such as nanog, oct4, TERT, SSEA-4, and GDF3 were not sufficient enough to correctly identify fully reprogrammed cells, as even partially reprogrammed cells could exhibit these features of pluripotency. Instead, the most reliable marker combination to identify fully reprogrammed cells were (1) retroviral silencing, (2) expression of Tra-1-60, DNMT3B, and Rex1, and (3) transient dim staining with Hoechst. The most stringent test of pluripotency is live organism production by tetraploid complementation and chimera formation. For obvious ethical reasons, these last two criteria cannot be employed for human cells (but see ref. 160, for chimera testing for human ESCs).
Molecular Analysis of iPSCs
Initial reports of iPSCs found them to be very similar to ESCs in terms of DNA methylation, gene expression, and other features related to pluripotency (361,362), but more detailed molecular analysis has suggested a distinct genetic signature that distinguishes iPSCs from ESCs (73,74,118,236) [but see refs. (124,267) for contrary]. Analysis of pluripotent-related gene expression, DNA methylation, cell cycle dynamics, microRNA, imprinted genes, and genomic stability has shown that iPSCs are indeed very similar to ESCs but also show some key differences (Table 3). Incomplete reprogramming or somatic memory may contribute to differences (18,118, 184,204,237,296). Interestingly, extended culture attenuates these differences, perhaps as iPSCs achieve a more fully pluripotent state, rendering late passage iPSCs transcriptionally closer to ESCs compared to early passage iPSCs (73,74,284,296,352). Exactly when this late stage is achieved seems to depend on the donor cell type (296). Parental cell origin or method of reprogramming does not seem to contribute to potential differences.
A Comparison of Pluripotent-Related Features of iPSCs and ESCs

iPSCs used for above data have passed all criteria (species appropriate) outlined in Table 2, (i.e., bona fide iPSCs). CNVs, copy number variation; Dlk1-Dio3, δ-like homologue 1-type 3 deiodinase; H19, imprinted maternally expressed long noncoding RNA transcript; KCNQOT1, imprinted paternally expressed long noncoding RNA transcript; lincRNA, long intergenic noncoding RNA; miRNA, microRNA; mtDNA, mitochondrial DNA; NNAT, neuronatin; ROS, reactive oxygen species.
Recent reports show that iPSCs seem to have increased genomic instability compared to ESCs (122). It is likely that the reprogramming process itself may be largely responsible for this (152,216,246), in addition to viral integration, amplification of exogenous transgenes, insertional mutagenesis (122,305,359), and incomplete reprogramming (somatic memory) (95,296). These aberrations may affect differentiation potential and increase tumor concerns. The reprogramming process may also compromise mitochondrial integrity (302). Importantly, a study analyzing mitochondrial DNA found that all iPSC clones analyzed maintained correct mitochondrial function and energy metabolism as ESCs regardless of low or high mutational load (302).
iPSC-Based Therapies as a Candidate for SCI Repair
Can iPSCs Form Functional Neurons? Neurite Induction Compared to ESCs
Although iPSCs share highly similar expression of genes related to pluripotency and development, there is evidence that iPSCs may occupy a distinct pluripotent “state” from ESCs (see “Molecular Analysis of iPSCs”). As such it is important to test that iPSCs have the same capacity as ESCs to generate a whole spectrum of region-specific neural progenitors and functional neuronal subtypes for SCI therapies (and other CNS disorders).
Since the initial isolation of ESCs, many neural induction protocols have been developed in attempts to yield functional neurons. Neural progenitors can be patterned along rostrocaudal and dorsoventral axes using specific morphogens such as sonic hedgehog, retinoic acid, fibroblast growth factor 8, or Wnt (for review, see ref. 129) in order to obtain region-specific neuronal subtypes such as spinal motor neurons, midbrain dopaminergic neurons, and forebrain glutamatergic and GABAergic neurons. Directed neural differentiation approaches in vitro generally involve either embryoid body (EB) formation, adherent monolayer-defined culture conditions, coculture with stromal feeder cells, or by default differentiation (348). Several optimizations in protocols have been shown to increase the yield of neural induction and include dual small body size–dual mothers against decapentaplegic (SMAD) inhibition with noggin and small molecule SB431532 (35,58,425), dual inhibition of activin/nodal and bone morphogenetic protein (BMP) signaling pathway (178) or sulfate reduction by chlorate treatment (323). Many studies have reported that iPSCs can be directed towards a neural lineage applying the same protocols (same developmental signals at the same concentration) that have been developed for ESCs (Table 4).
General Neural Induction in iPSCs—Mouse Donor Cells
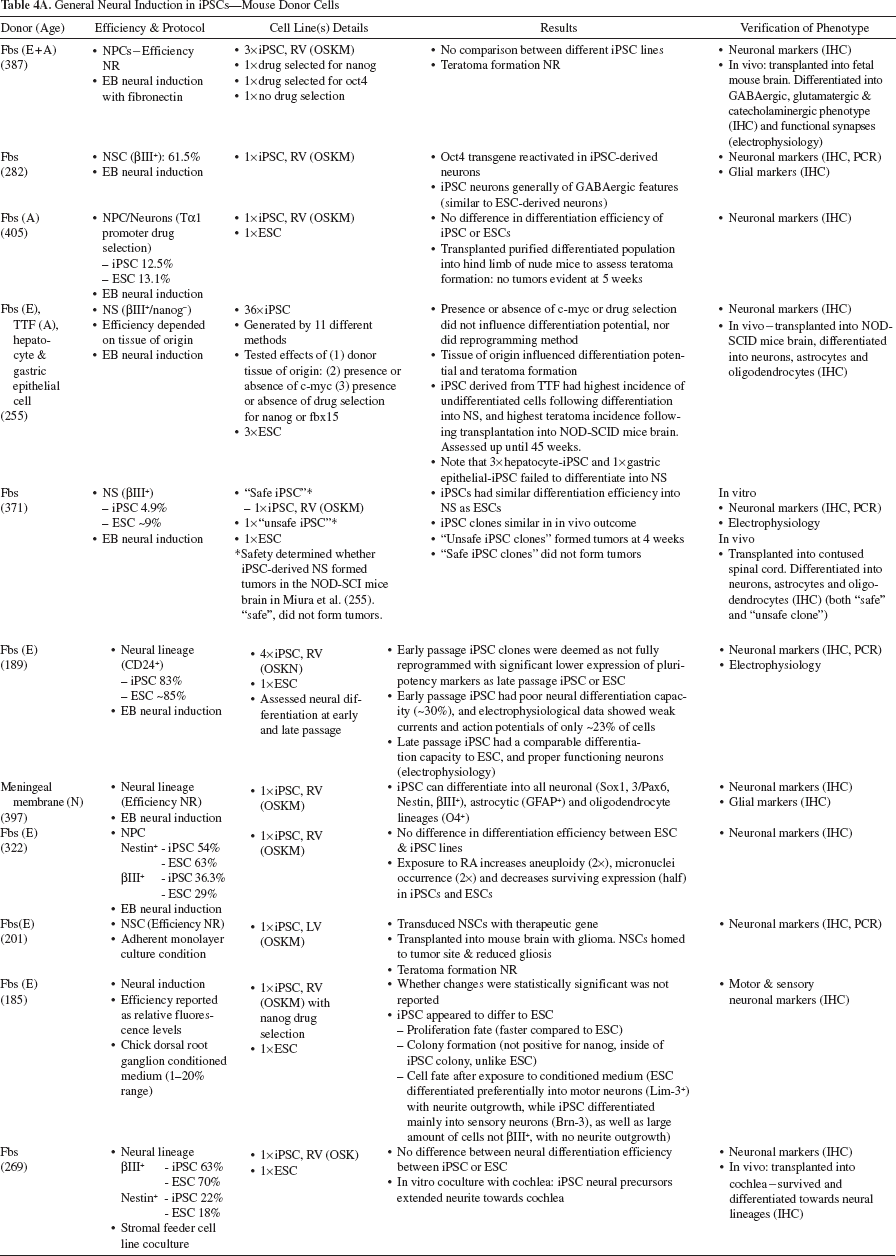
General Neural Induction in iPSCs—Human Donor Cells
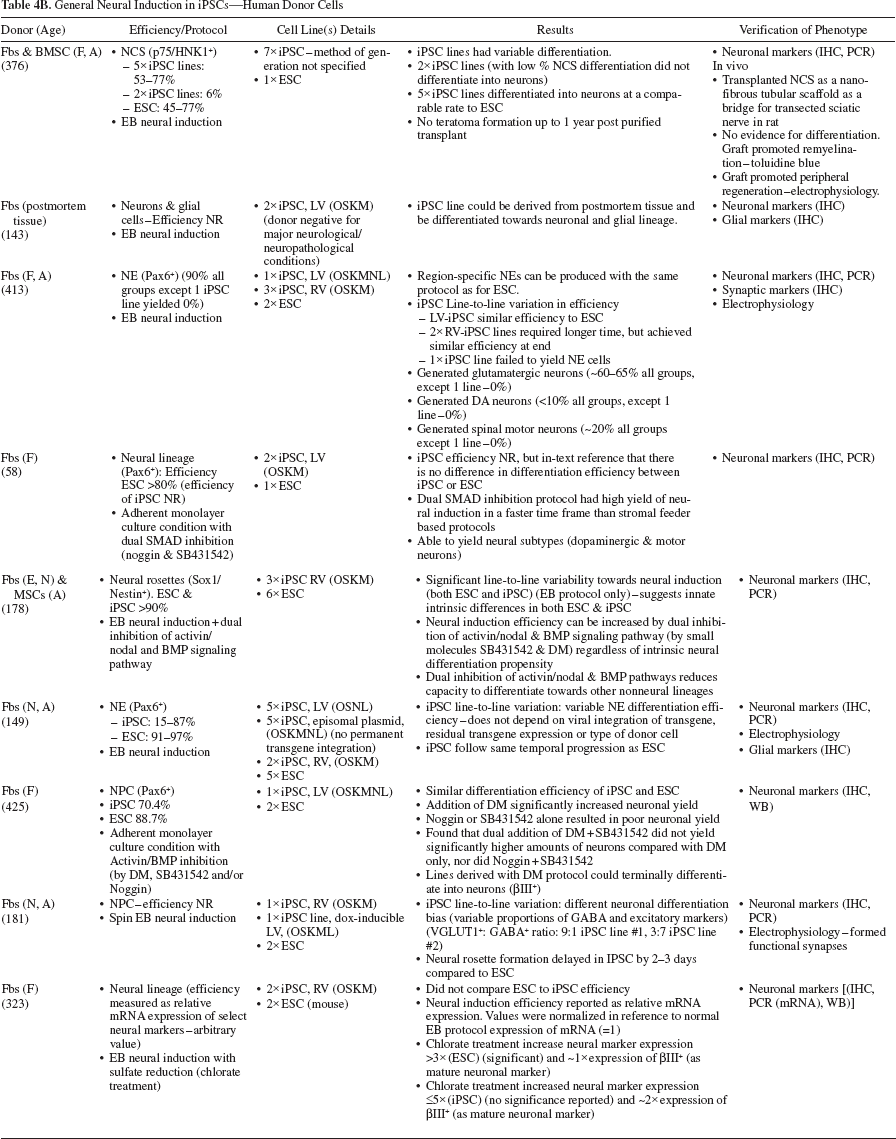
Specific Neuronal Subtype Induction—Midbrain Dopaminergic Neurons

Specific Neuronal Subtype Induction—Motor Neurons

Neural Induction—iPSC Derived From Donors With Neurological Disease

Induction Towards Oligodendrocyte Lineage

Induction Towards Astrocytic Lineage

Induction Towards Retinal Neurons
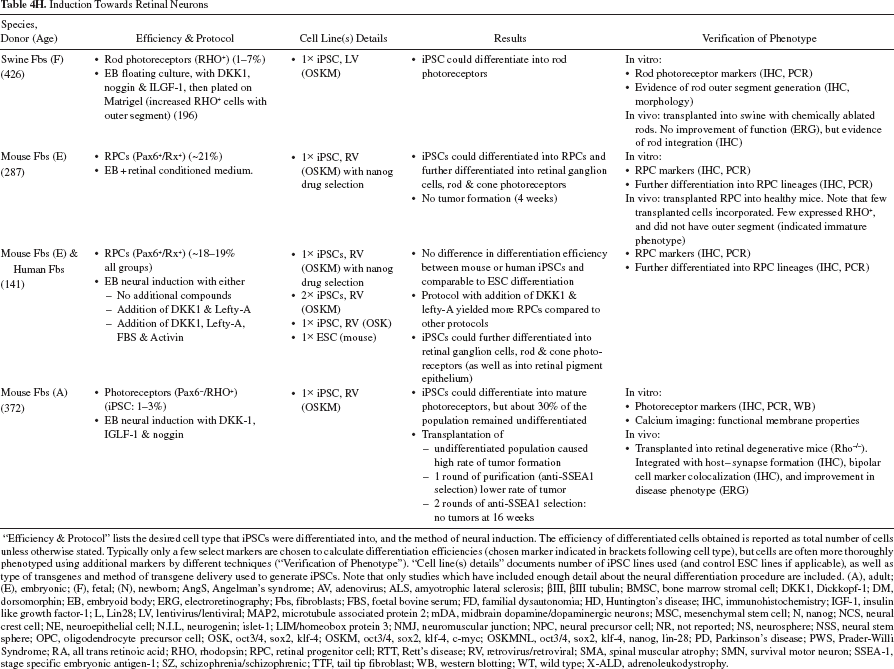
“Efficiency & Protocol” lists the desired cell type that iPSCs were differentiated into, and the method of neural induction. The efficiency of differentiated cells obtained is reported as total number of cells unless otherwise stated. Typically only a few select markers are chosen to calculate differentiation efficiencies (chosen marker indicated in brackets following cell type), but cells are often more thoroughly phenotyped using additional markers by different techniques (“Verification of Phenotype”). “Cell line(s) details” documents number of iPSC lines used (and control ESC lines if applicable), as well as type of transgenes and method of transgene delivery used to generate iPSCs. Note that only studies which have included enough detail about the neural differentiation procedure are included. (A), adult; (E), embryonic; (F), fetal; (N), newborn; AngS, Angelman's syndrome; AV, adenovirus; ALS, amyotrophic lateral sclerosis; βIII, βIII tubulin; BMSC, bone marrow stromal cell; DKK1, Dickkopf-1; DM, dorsomorphin; EB, embryoid body; ERG, electroretinography; Fbs, fibroblasts; FBS, foetal bovine serum; FD, familial dysautonomia; HD, Huntington's disease; IHC, immunohistochemistry; IGF-1, insulin like growth factor-1; L, Lin28; LV, lentivirus/lentiviral; MAP2, microtubule associated protein 2; mDA, midbrain dopamine/dopaminergic neurons; MSC, mesenchymal stem cell; N, nanog; NCS, neural crest cell; NE, neuroepithelial cell; N.I.L, neurogenin; islet-1; LIM/homeobox protein 3; NMJ, neuromuscular junction; NPC, neural precursor cell; NR, not reported; NS, neurosphere; NSS, neural stem sphere; OPC, oligodendrocyte precursor cell; OSK, oct3/4, sox2, klf-4; OSKM, oct3/4, sox2, klf-4, c-myc; OSKMNL, oct3/4, sox2, klf-4, nanog, lin-28; PD, Parkinson's disease; PWS, Prader-Willi Syndrome; RA, all trans retinoic acid; RHO, rhodopsin; RPC, retinal progenitor cell; RTT, Rett's disease; RV, retrovirus/retroviral; SMA, spinal muscular atrophy; SMN, survival motor neuron; SSEA-1, stage specific embryonic antigen-1; SZ, schizophrenia/schizophrenic; TTF, tail tip fibroblast; WB, western blotting; WT, wild type; X-ALD, adrenoleukodystrophy.
To date, iPSCs have been directed to generate neural crest cells (201,376), peripheral sensory neurons (185), neural stem cells, and their neuronal progenitors including specific neuronal subtypes such as glutamatergic (181,238,292,413), GABAergic (42,178,238), dopaminergic neurons (Table 4C), motor neurons (Table 4D), as well as retinal neurons (Table 4H), oligodendrocytes (Table 4F), and astrocyte lineages (Table 4G). iPSCs follow the same temporal progression of differentiation as ESCs and can also be patterned into region-specific neuronal subphenotypes with the same morphogens. Similar to ESCs, iPSC-derived neurons differentiate into default anterior phenotype unless morphogens are applied to obtain a posterior phenotype (181,192,413). Variations in some iPSC lines have been noted; different lines have enriched dorsal marker expression (181) and may show differential bias towards GABAergic or excitatory neurons (181,282). Also of note, neural rosette formation has been found to be delayed for 2–3 days in some human iPSC lines compared to their human ESC counterpart (178), as well as produce a lower yield of neural specific EB (172). Nevertheless, directed neural differentiation of iPSCs can result in electrophysiologically active neurons that are able to form functional synapses (35,42,138,149,172,181,189,238,268,292,307,321,371,376,387,413).
Some authors have reported differences in the differentiated efficiency between iPSCs and ESCs as well as differences between the various iPSC lines themselves (35, 91,101,149,178,255,368,371,376,413) and between different ESC lines (178,283). On the other hand, other studies have not detected such differences between iPSC lines or ESCs (42,49,58,89,100,132,161,181,189,192,238,268,275, 325,338,358,387,416,425). Variation in differentiation efficiency is thought to reflect innate intrinsic differences of individual iPSCs (and ESCs). Some factors that have been identified to contribute to differences in efficiency are donor cell sex and parental donor cell origin (35,255), which may represent unique genetic composition favoring certain cell fates over others. Partially reprogrammed iPSCs are less likely to differentiate. It has been reported that early passage iPSCs may occupy a less fully reprogrammed state than late passage iPSC, which may differentiate more efficiently into neurons, as well as have improved excitability and increased voltage-gated currents (189). Yet Boulting et al. (35) observed no such differences between late and early passage numbers. Factors that are not thought to influence iPSC differentiation propensity include the method of reprogramming (number and choice of transcription factors used, choice of transgene delivery, and presence or absence of drug selection for iPSC clones) (35,149,255,338,387) [note that recently a link between differentiation efficiency and number of reprogramming factors has been reported (221)], donor age (35), donor health status (Table 4E), or whether iPSC clones have permanent genomic integration of transgenes or transient transgene expression only (35,149,234,338). iPSC clones that have reactivated exogenous transgenes, such as c-myc, are likely to have impaired differentiation potential as c-myc is well known to inhibit differentiation and promote cell proliferation (11,334,339). Tables 4A–H summarize the current knowledge of specific neuronal subtype induction from iPSCs, detailing donor cells, protocol, and efficiency, as well as assessment of phenotype.
iPSCs in Spinal Cord Injury and other In Vivo Models
To date, iPSCs and their derivatives have been used in various in vivo animal models of neurological/neurodegenerative disorders including SCI (Table 5), Parkinson's disease (49,89,132,307,321,387), demyelination (85,299), retinal regeneration (287,372), stroke (66,164,402), and peripheral nerve regeneration (376), as well as being used in several nonneurological models such as liver disease, cardiac infarction, hind limb ischemia, anemia, hemophilia, bone regeneration, and tracheal defect (Table 6). These studies have provided a proof of principle that iPSCs can be successfully differentiated in vitro to yield desirable progeny and which can be effectively subjected to ex-vivo gene therapy (131,396). iPSCs transplanted into host tissue can survive, integrate, differentiate into desired phenotypes and attenuate disease phenotype with a comparable outcome to the ESC counterpart therapy.
iPSC-Based Therapies in Experimental SCI

5HT, serotonin/serotonergic; APC, adenomatous polyposis coli; βIII, βIII tubulin; BBB-LS, Basso, Beattie, Bresnahan locomotion scale; BMS, Basso mouse scale; CBR/uc, click beetle red luciferase; CGRP, calcitonin gene related peptide; DMEM, Dulbecco's modified Eagle's medium; ECM, extracellular matrix; ESC, embryonic stem cells; Fbs, fibroblasts; GAP43, growth associated protein 43; GFAP, glial fibrillary acidic protein; π-GST, glutathione-S-transferase-pi; IH, infinite horizon; IHC, immunohistochemistry; kd, kilo dynes; LFB, Luxul fast blue; MBP, myelin basic protein; MEF, mouse embryonic fibroblast; NeuN, neuronal nuclei; NF, neurofilament; NOD-SCID, non-obese diabetic severe combined immunodeficient; NS, neurospheres; OSKM, Oct4, sox2, klf4, c-myc; PBS, phosphate buffered saline; PECAM-1, platelet endothelial cell adhesion molecule-1; PNS, primary neurospheres; RV, retrovirus; SCI, spinal cord injury; SNS, secondary neurospheres; TTF, tail tip fibroblast; VEGF, vascular endothelial growth factor.
Base of support: average width between front legs (or hind legs).
Placement of feet: position of the hind foot in relation to the front foot (on the same side).
Safety was determined by transplanted iPSC-SNS into NOD-SCID mice brain–clones that formed tumors were designated “unsafe”(255).
Summary of iPSC-Based Strategies in Both CNS In Vivo Models (Stroke & Demyelination) and Nonneuronal In Vivo Models to Date
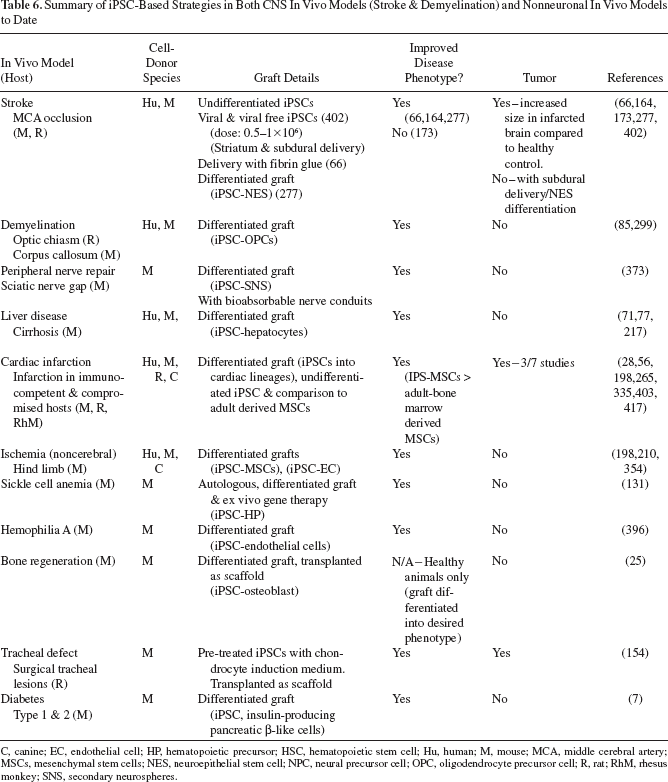
C, canine; EC, endothelial cell; HP, hematopoietic precursor; HSC, hematopoietic stem cell; Hu, human; M, mouse; MCA, middle cerebral artery; MSCs, mesenchymal stem cells; NES, neuroepithelial stem cell; NPC, neural precursor cell; OPC, oligodendrocyte precursor cell; R, rat; RhM, rhesus monkey; SNS, secondary neurospheres.
Currently, there are only a few published studies describing the use of iPSCs in SCI therapies (Table 5). All published groups reported on the incidence of neuronal, glial, and astrocytic marker expression after transplantation, with two groups reporting differentiation of donor cells into these various cell types (271,371). These studies used iPSC donor cells that were differentiated into either neurospheres (NS) (271,371) or astrocytes (136). Our own unpublished studies using undifferentiated human iPSCs injected into a nude rat model of acute moderate contusion SCI are included. It is important to consider that appropriate cell controls should be used in preclinical studies to evaluate the possibility of small populations of incompletely reprogrammed donor cells and/or incompletely predifferentiated donor iPSCs contributing to the morphological and functional outcomes observed after treatment. Our studies using undifferentiated iPSCs (from human fibroblast donor) as well as human dermal fibroblast controls effectively showed no significant differences in morphological or functional outcomes by 5 weeks after SCI, despite subtle differences in some neuronal marker expression at the lesion site. Similarly, Hayashi et al. (136) reported no significant differences in morphological or functional outcomes in another acute moderate contusion SCI model in rats. While these studies may suggest that improved outcomes were observed in mouse but not in rat models of SCI, the disparity in overall results from these very limited number of studies suggest that iPSC-based therapy in SCI warrants more extensive and thorough testing. Ideally, research in this area should be conducted using comparable regimes in mouse and rat models, as outlined in the recommendations and guidelines developed by the International Campaign for Cures of Spinal Cord Injury Paralysis (ICCP) (107).
As with pluripotent ESCs, several iPSC in vivo studies have reported high rates of teratoma formation (12,21,49–51,63,66,255,272,307,319,357,371,372,383,402,405), a risk that seems to be increased with (i) c-myc or klf-4 reactivation (279), (ii) higher dosage of cells delivered (272,335,403) (also see ref. 417), (iii) changes in local environment; necrotic tissue such as the infarcted myocardium seems to promote tumor expansion (402) (importantly, in SCI necrotic tissue is abundant), and (iv) the amount of undifferentiated cells present in the graft (85,255, 371,372). This latter point can be easily addressed by purifying the population by flow cytometry (e.g., negative selection for SSEA-1+ cells), which can prevent tumor formation (372,387). But other studies have reported no evidence of tumor formation in various animal models assessed at time points ranging from as early as 5 weeks to as late as 1 year (85,89,132,136,234,358,376,405). Of note, although one of the major advantages of iPSCs is potential patient-specific cells for treatment of disease, only very few studies to date have used autologous iPSCs for transplantation (131,198), which is important as recent evidence suggests that iPSCs may evoke an immune response in syngeneic recipients (419) (see “Immunogenicity of Pluripotent Stem Cells”).
Issues with Using iPSC-Based Therapies
Tumorigenicity of iPSCs
Pluripotent stem cells and tumors share characteristics such as proliferative self-renewal, lack of contact inhibition and telomerase activity, and they share expression of many common genes (29,75,117,341,393,415), and it has been shown that both pluripotent stem cells and their ESC derivatives can form teratomas in vivo (see “iPSCs in Spinal Cord Injury and Other In Vivo Models”). Teratoma formation is thought to be strongly linked to the number of undifferentiated cells in the graft (21,272). Furthermore, in vitro culture may inadvertently select for more tumorigenic ESCs/iPSCs, because clones that are fast growing and less likely to spontaneously differentiate are more likely to be preferentially propagated. These characteristics make the pluripotent stem cell likely to be more tumorigenic in vivo (187). Recent reports have indicated that iPSCs have increases risks of tumorigenicity compared to ESCs, as reprogramming factors used to induce pluripotency are oncogenic (c-myc and klf-4) (279), or linked to tumor pathways, and differences between iPSCs and ESCs in gene expression, epigenetics, and genomic instability, which have been linked to increased tumorigenicity [for reviews, see refs. (23,187,191,258,276,391)]. Indeed, injection of iPSCs into nonobese diabetic/severe combined immunodeficient/interleukin 2γ-null (NOD/SCID/IL-2-/-) mice (with both adaptive and innate immune deficiencies) resulted in aggressive teratomas with higher frequency and reduced time of onset compared to ESCs (126).
Tumor formation risk can however be reduced with several strategies (for review, see ref. 187). Terminal differentiation of ESCs/iPSCs reduces the risk of teratoma formation, but differentiation alone cannot completely eliminate pluripotent stem cells from the population (113). Sorting by flow cytometry, for example, with negative selection of SSEA-1, which should be performed at least twice (372), antibody depletion of SSEA-5 (363), or possibly by natural killer (NK) cell targeting of pluripotent stem cells (97). The methods of obtaining iPSCs can also be modified to reduce the risk of tumors; the use of l-myc instead of c-myc (see “Generation of iPSCs: General Considerations of Delivery Approaches”), the use of nonintegrating transgene delivery systems to prevent tumorigenesis from reactivation of transgenes and/ or the integration of a suicide gene (50,68,200,423). Rigorous analysis of the cell line to select the lines with the least propensity of tumor formation for transplantation may also be beneficial (11,255,371). Lastly, regulation of glycogen synthase kinase 3β (Gsk3β) pathway may also be beneficial in reducing the tumor formation, at least in ESCs (209).
Immunogenicity of Pluripotent Stem Cells
ESCs have demonstrated immune-privileged properties such as low levels of human leukocyte antigen (HLA) class I, absence of HLA class II, reduced NK ligands, as well as several in vivo demonstrations of immune evasion by transforming growth factor-β2 and serpin-6 expression, inducing T-cell apoptosis and T-regulatory-mediated suppression, in syngeneic and allogeneic transplants (3,31,98,99,188,207), even across a full major histocompatibility complex (MHC) barrier (226). But it is now becoming increasingly clear that ESCs are not truly immune privileged with several studies reporting that ESCs are capable of eliciting strong immune responses from both T-cells and NK cells (in syngeneic, allogeneic and xenogeneic recipients) (38,97,98,190,357). Inducing tolerance can be done with relative ease (in comparison to adult derived tissue), for example, by costimulatory or coreceptor blockage (37,105,226,311). In contrast, the immunogenicity of iPSCs has not been extensively investigated so far. To date only four seminal reports on the immunogenicity of iPSCs have been published (89,97,291,419) (Table 7), and there are too few data to draw definite conclusions on iPSC immunogenicity. However, it is becoming clear that differences between iPSC and ESC immunogenicity may exist and it should not be simply assumed that an iPSC autograft will not be rejected by the host immune system.
Summary of Reports Investigating Immunogenicity of iPSCs

LAK, lymphocyte activated killer cell; NK, natural killer (cell).
Immunoregulation by Donor Cells
The potential immunoregulatory capacity of many donor stem cell populations may influence many factors posttransplantation, including donor cell survival, successful engraftment (and subsequent transdifferentiation capability?) within the required site, as well as induction of endogenous (host) stem cell and/or repair mechanisms. Crosstalk between implanted donor stem cells and recipient immune cells (220) may therefore have a key role in determining the success of stem cell-mediated tissue regeneration (32). Mechanisms may involve inhibition of specific immune cell proliferation, activation, induction of tolerance (370), and/or suppression of maturation and function of specific subsets of populations (6). It is likely that donor iPSC transplantation may also involve interplay between these potential immunoregulatory factors in the host.
Tracking the Fate of Donor iPSCs
Accurate assessment of the fate of donor cells in transplantation therapies often requires the use of markers to monitor the fate of grafted cells and any influence of/interaction with these cells on the host. Many types of protocols that have been used have their advantages and disadvantages, including immunohistochemistry (IHC), retrograde-labeling, electron microscopy (EM), and fluorescence in situ hybridization (FISH) (133). Technical limitations can also be compounded by factors such as immunogenicity of the marker of choice leading to specific immune responses (347) and ambiguity in assessment due to (transient?) loss of long-term expression of the marker in surviving cells (146,203,256), leakage of marker and subsequent uptake by host cells, toxicity from chronic exposure of markers, change in cellular distribution over time, and uptake/redistribution by scavenging host microglia/macrophages. Other markers may circumvent these factors by the use of species-specific markers, markers associated with a specific gene promoter or adenoviral (AdV), adeno-associated viral (AAV), retroviral (RV), and lentiviral (LV) vectors for the introduction of foreign genes encoding either the bacterial marker enzyme β-galactosidase/LacZ or green fluorescent protein (GFP), fluorescent (usually rhodamine or fluorescein) dye-coated latex microspheres/beads/nanospheres, and combined analysis with other assays such as IHC, retrograde-labeling, or EM, FISH, or real-time quantitative PCR using donor-specific sequences [e.g., Y1 DNA of male cells transplanted into female hosts (34,134), as well as the use of thymidine, bromodeoxyuridine (BrdU), and ethynyl deoxyuridine (EDU) analogues (133)].
Conclusion
iPSCs are a novel source of neural stem cells for SCI therapy that do not share the disadvantages of adult or embryonic-derived cells. To date, only a few studies have investigated iPSC-derived grafts in SCI models. The disparity in overall results allows no clear conclusions to be drawn as to how iPSC-based therapy compares to cells derived from other sources. It is important to be mindful of certain factors (e.g., choice of donor cell or method of transgene delivery) that may affect iPSC differentiation propensity towards neural lineage and may ultimately also affect in vivo outcome. As such, future in vivo studies should carefully consider (and provide sufficient details) on their method of choice for iPSC generation and characterization to ensure that only authentic iPSCs are used. At present, most in vivo studies to not provide sufficient details regarding the iPSCs used for transplantation, which can limit the conclusions that can be drawn from these reports
Despite the promise iPSC technology holds in providing an unlimited source of desired donor cells, current technologies appear to be too inefficient, costly, and time-consuming, especially for safer nonviral, non-DNA-integrating methods, to be useful in a clinical setting anytime soon. Protocols must be continually developed to address these issues. Furthermore, after establishment of a new patient-specific cell line, it must be ensured that (i) the cell line is appropriate for the establishment of the necessary cells (line to line variability in establishment of tissue) and (ii) the donor population is devoid of undifferentiated cells and does not form tumors in vivo. At present, these processes can take several months to complete, which is not ideal in SCI since acute transplants (and other noncellular therapies) are vastly superior in functional and morphological improvements compared to chronic injury regimes, which tend to show greatly diminished, if any, improvements (148). Our understanding of how to direct differentiation towards a potentially heterogeneous population of donor cells that incorporate more stringent safety features also needs to be improved. Lastly, from preliminary investigations into the immunogenicity of iPSCs, it is becoming apparent that iPSC may be targeted by the immune system, requiring immune suppression/tolerance induction regimes. For these reasons, it is more likely that—with the current technologies and knowledge—an iPSC bank with ready-made cells produced under the most stringent GMP conditions, appropriately evaluated and characterized, will be more feasible than creating new patient-specific lines for the urgent treatment of SCI and other types of neurotrauma.