
Abstract
Phenylketonuria is a metabolic disease caused by phenylalanine hydroxylase deficiency. Treatment is based on a strict natural protein-restricted diet that is associated with the risk of malnutrition and severe psychosocial burden. Oral administration of tetrahydrobiopterin can increase residual enzyme activity, but most patients with severe clinical phenotypes are nonresponders. We performed liver cell transplantation in a 6-year-old boy with severe tetrahydrobiopterin nonresponsive phenylketonuria who failed to comply with diet prescriptions. The transplanted hepatocytes were obtained in part from an explanted glycogen storage type 1b liver. Following two infusions, blood phenylalanine levels returned within the therapeutic target while the phenylalanine half-life assessed by loading tests decreased from 43 to 19 h. However, 3 months later, blood phenylalanine concentrations increased and the phenylalanine intake had to be reduced. Cell-based therapy is a promising therapeutic option in phenylketonuria, and the domino concept may solve the issue of cell sources for hepatocyte transplantation.
Introduction
Phenylketonuria (PKU) is one of the most common inherited metabolic diseases, caused in most cases by deficiency of the phenylalanine hydroxylase (PAH) enzyme in the liver. The clinical phenotype ranges from classic PKU to mild hyperphenylalaninemia with increasing residual enzyme activity (17). Long-term cognitive outcome is correlated with the mean blood phenylalanine concentration during infancy and a meta-analysis established a 1.9-to 4.1-point reduction in intelligence quotient for each 100 μmol/L increase in the mean lifetime phenylalanine concentration (2, 10, 19, 23). Since more than half a century, the treatment of PKU relies on a strict dietary natural protein restriction in order to maintain blood phenylalanine under target concentration and prevent neurotoxicity of hyperphenylalaninemia and associated long-term cognitive impairment. This highly restrictive diet represents a severe burden for patients and families and is responsible for a substantial impairment of the patient's quality of life (2). Moreover, a patient following such a modified diet is at risk for nutritional deficiencies. Finally, intermittent metabolic imbalances caused by recurrent catabolic states during minor infectious illness and occasional loss of compliance account for a residual risk of long-term intellectual disabilities, especially for patients with more severe phenotypes. Moreover, adult patients with PKU display behavioral disorders and PKU mothers may intoxicate their offspring during pregnancy (8).
Searching for nondietary treatment alternatives, tetrahydrobiopterin (BH4), the natural cofactor of the enzyme, has recently emerged as a major therapeutic option. It leads to correction of the biochemical phenotype by increasing the residual phenylalanine hydroxylase enzyme activity and, consequently, the dietary phenylalanine tolerance (11). However, only 20–25% of patients benefit from tetrahydrobiopterin, whereas most patients with severe clinical phenotypes due to mutations associated with no residual enzyme activity are nonresponders (1). Other alternative treatment strategies currently under clinical or preclinical development include enzyme replacement therapy by subcutaneous administration of phenylalanine ammonia lyase and gene therapy (15, 16). Clinical trials with enzyme replacement therapy are underway, and gene therapy remains a promising approach but not yet clinically applied.
The metabolism of phenylalanine is mainly liver based. Hence, liver cell transplantation is an attractive option to help controlling phenylalanine levels in the most severely affected patients. Hepatocyte transplantation has been shown to restore missing liver enzyme deficiencies at a degree to modify the clinical course of severe liver based metabolic diseases such as Crigler–Najjar, urea cycle deficiencies, glycogen storage diseases, Refsum disease, and factor VII deficiencies (18). Since organ shortage limits access to hepatocytes for cell transplantation, the use of the domino technique may in future be an appealing alternative to increase the number of treated patients.
Patients and Methods
Patient
The patient was a 6-year-old boy with classic PKU diagnosed in the neonatal period by the newborn screening program (initial phenylalanine concentration before treatment: 3,120 μmol/L). He was compound heterozygote for the P281L and IVS10-11G>A mutations in the PAH gene. According to the PAH database Web site (www.pahdb.mcgill.ca), both mutations are considered as null mutations with no residual enzyme activity and thus responsible for a severe phenotype. Tetrahydrobiopterin responsiveness has been ruled out by a loading test with 20 mg/kg of sapropterine dihydrochloride (Kuvan®, Merck) over a period of 24 h (3). Dietary surveys determined the phenylalanine tolerance of the patient to be 250 mg/day, representing the oral phenylalanine intake allowing to maintain blood phenylalanine concentrations of <360 μmol/L, the most consensual therapeutic target concentration (1).
Although good metabolic control was achieved in the first 2 years of life, his metabolic state progressively deteriorated after the age of 2 years due to poor compliance with the diet and despite a close medical, dietary, and psychosocial follow-up in a specialized metabolic unit agreed for PKU management by the Federal Ministry of Health. The child had frequently elevated levels of phenylalanine above the safe limit of 360 μmol/L. During the first 2 years of life, between 2 and 4 years and between 4 and 6 years, mean blood phenylalanine concentrations were 228 ± 132 μmol/L (n = 93), 444 ± 312 μmol/L (n = 23), and 588 ± 264 μmol/L (n = 21), respectively. The child was referred for hepatocyte transplantation in order to improve his metabolic control and aiming to transform his severe biochemical phenotype into an easier manageable form of the disease. The institution performing the procedure has an ongoing clinical protocol of liver cell transplantation approved by the institution review board and has also an approved cell isolation facility within an agreed Tissue Bank, allowing for the preparation of clinical grade hepatocytes for transplantation. The indication for liver cell transplantation was accepted by the multi-disciplinary team on the basis of the medical history and lack of control of the disease despite a specialized close follow-up of the child and family. It was approved by the institution's ethical review board (Saint-Luc University Hospital), and a written informed consent from the parents was obtained.
A pretransplant evaluation was performed, and any associated condition, which would be a contraindication to the procedure, was excluded. In particular, patency of the portal system, absence of portosystemic shunting, and normal echo structure of the liver parenchyma were verified. Blood group of the patient was A Rhesus positive.
First Series of Infusions
The child was admitted to the hospital the day prior to the infusion. A transcutaneous portal catheter was placed under combined Doppler ultrasound and angiographic guidance in the right portal vein, and the extremity was advanced to the splenomesaraic confluent. Final position and patency of the catheter was controlled by fluoroscopy. The child was then kept under general sedation in the paediatric intensive care unit until the end of the infusion period, for a total of 18 h. Catheter position was controlled before each infusion by Doppler ultrasound, and blood flow velocity in the portal system was measured. Portal pressure was also monitored before and after each infusion.
The cells injected came from the explanted liver of a 14-month-old female patient with type Ib glycogen storage disease (A Rhesus positive) undergoing liver transplantation from a living donor. Liver isolation yield was 3.5 billion cells with more than 90% of viability as estimated by the Trypan Blue test. The domino procedure was proposed by the surgical team and accepted by the institution's ethical review board in view of the liver donor shortage and lack of priority given for cell transplantation. The donor had no evidence of liver adenoma. Cells were infused in albumin 5% suspension, with 5 mM N-acetyl-l-cystein and 1 IU/ml heparin, at a rate of 60 ml/h of the suspension containing 10 × 106 cells/ml, as previously reported. The patient received two infusions for a total of 630 × 106 cells with 82% viability on day 1 evening and two injections for a total of 1.1 × 109 cells with 78% viability on day 2. The portal flow remained normal, with normal flow velocities of 20 cm/s in the portal vein branches and 30 cm/s in the main portal vein. The child experienced one episode of tremor, while his body temperature had dropped to 34°C, after starting the second infusion with cells preserved on wet ice, attributed to insufficient warming of the cell suspension before infusion. After the last infusion on day 2 and control of the Doppler ultrasound, the catheter was removed and the child woke up. Subcutaneous calcium nadroparine (Fraxiparine®, GSK, Belgium) was then started in view of a moderately elevated D-dimers level of 3,300 ng/ml (normal <500) at the end of the four infusions, aiming to dissolve possible small peripheral microthrombi.
The patient was maintained under tacrolimus (Prograft®, Astella, Belgium) aiming to reach a blood level of 6–8 ng/ml. He received 1 mg/kg/day of prednisolone during the infusions. The child was discharged the next day.
Second Series of Infusions
Seven and a half months later, the child received a last infusion of 850 × 106 freshly isolated hepatocytes from a 48-year-old cadaveric male donor 0 Rhesus positive (cerebral trauma). Liver isolation yield was 3.4 × 109 cells with more than 82% of viability as estimated by the Trypan Blue test. The infusion was again made via a percutaneous catheter over 1 h in one injection. He was maintained under tacrolimus aiming to reach blood levels of 6–8 ng/ml.
Posttransplantation Evaluation
The same diet prescription was maintained before and after the transplantations.
Biochemical Monitoring
Blood phenylalanine levels (μmol/L) were monitored each week posttransplantation by ion exchange chromatography (Biochrom 30). Liver function tests and α-fetoprotein levels were also routinely followed up.
Liver Biopsies
Liver biopsies were performed 3 months after each infusion to determine PAH tissue activity. The PAH activity was evaluated on hepatic biopsies by conversion of phenylalanine into tyrosine (pmol/min/mg of tissue). As a control, PAH activity was measured in a liver sample of a donor with normal PAH activity.
Phenylalanine Elimination Kinetic Analyses
A phenylalanine loading test was performed before and after transplantation. The patient received 60 mg of l-phenylalanine per kilogram of body weight in a single oral dose. Blood phenylalanine concentrations were measured by ion exchange chromatography (Biochrom 30) before and 40 min, 80 min, 2 h, 3 h, 4 h, 6 h, 8 h, 10 h, and 12 h after loading. During this period, the patient received his usual standardized diet containing 250 mg of phenylalanine a day. The maximal phenylalanine level was considered as the first point of the phenylalanine disposal curve. In this kinetic model analysis, phenylalanine decline is described as a single exponential decay with the half-life t1/2 = –(ln2)/slope (3, 7). Kinetic analysis was performed using the EnzFitter software (Biosoft, Cambridge, UK) with the Marquardt–Levenberg algorithm in view to minimize errors from outliers in the nonlinear process.
Statistics
The p values compare blood phenylalanine concentration using a Mann–Whitney two-tailed test. The significant values were adjusted according to Bonferroni correction to avoid type 1 error.
In the kinetic model analysis, phenylalanine decline is described as a single exponential decay with the half-life t1/2 = –(ln2)/slope (3, 7). Kinetic analysis was performed using the EnzFitter software (Biosoft, Cambridge, UK) with the Marquardt–Levenberg algorithm in view to minimize errors from outliers in the nonlinear process.
Results
Safety of the Infusions
Liver function tests [aspartate aminotransferase (AST) and alanine aminotransferase (ALT)] slightly increased during the infusion periods (maximum AST value 479 UI/ ml and maximum ALT value 254 UI/ml before normalization within 2–3 days. α-Fetoprotein levels remained within normal values throughout the study period (data not shown).
Effects of Hepatocyte Transplantation on Blood Phenylalanine Concentration, Phenylalanine Disposal, and Oral Phenylalanine Tolerance
Blood phenylalanine concentrations in the 12 months preceding the first transplantation, between the two transplantations, and after the second transplantation are shown in Figure 1. A clinically significant effect was clearly observed after the second hepatocyte infusion, where phenylalanine levels reached target concentrations whereas no change had been prescribed in dietary intakes. Three weeks after the second transplantation, phenylalanine concentrations decreased below the recommended cutoff for this essential amino acid (<60 μmol/L) and natural protein intake had to be increased from 5 to 9 g/ day, in view to avoid phenylalanine deficiency. Daily oral phenylalanine tolerance, assessed after the second transplantation by objective measurement of the phenylalanine intakes during hospitalization, showed an increase from 250 mg/day before transplantation to 450 mg/day after transplantation. Overall, these results indicate that the high phenylalanine concentrations observed before transplantations were caused by noncompliance with the diet prescriptions, the patient most probably eating more than 400 mg of phenylalanine per day. After the second transplantation, owing to improvement of his tolerance, a dietary intake of 450 mg of phenylalanine per day allowed to keep his blood phenylalanine concentration of <360 μmol/L. However, this beneficial effect indicating an increase of in vivo PAH activity was limited to a time period of 3 months (Fig. 1).
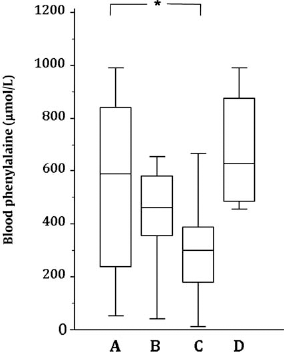
Effect of hepatocyte transplantations on blood phenylalanine concentrations. Blood phenylalanine concentrations (A) in the 12 months before hepatocyte transplantation (n = 18); (B) from 1 month after the first transplantation until second transplantation (n = 25); (C) in the 3 months following the second transplantation (n = 23); (D) after 3 months following the second transplantation (n = 12). The boxes represent the interquartile ranges, the internal bars are the medians, and the external bars indicate the range. n, number of analyses, *p < 0.05.
In addition, we evaluated the success of the cell transplantation by performing phenylalanine loading tests. These were carried out before transplantation, 3 months after the first transplantation, and 6 weeks after the second transplantation. Results are shown in Figure 2. After the first transplantation, despite higher phenylalanine concentrations related to higher phenylalanine levels at the start of the test, the phenylalanine half-life assessed by kinetic analysis showed a mild reduction from 43 to 34 h. However, this degree of reduction was not considered to be clearly clinically significant, leading to the decision for a second intraportal infusion. The phenylalanine elimination curve after the second transplantation showed a strong reduction of phenylalanine half-life to 19 h, which was consistent with the overall improvement of the metabolic control of the patient.

Effect of hepatocyte transplantations on phenylalanine disposal after oral loading test. Evolution of blood phenylalanine concentration before (solid line), after the first transplantation (short dashed line), and after the second transplantation (long dashed line). Phenylalanine half-lives before, after the first, and after the second transplantation were 43, 34, and 19 h, respectively.
Phenylalanine Hydroxylase Activity in Liver
PAH activity was evaluated on frozen liver biopsies. A pretransplant biopsy was not available, since the patient had a genetically proven severe form of the disease known to display no residual activity.
Liver biopsies were performed 3 months after each infusion (11 months post-first transplantation for the second biopsy). Posttransplant PAH activity was measured at 13% and 6.6% when compared with a healthy control, respectively (Fig. 3).

Phenylalanine hydroxylase (PAH) activity (pmol/ min/mg of tissue) on posttransplantation liver biopsies evaluated by conversion of phenylalanine into tyrosine.
Discussion
Our report demonstrates that hepatocyte transplantation can transform severe PKU into a mild phenotype of hyperphenylalaninemia and opens the field of cell therapy for the management of PKU patients. Indeed, besides clinical improvement, normal blood phenylalanine levels were obtained after the second hepatocyte transplantation, probably due to a cumulative effect of the infusions. This was correlated with a detectable liver enzyme activity (13% and 6.6% of normal control activity, respectively, before and after the second transplantation). A lower enzyme activity on the post-second transplantation biopsy can probably be explained by sampling phenomenom. We know from animal studies, transplantation of PAH-positive hepatocytes into PAH-deficient Pahenu2 mice, that a significant decrease in serum phenylalanine is already observed when liver repopulation exceeded approximately only 5% (4). However, the metabolic effect of hepatocyte transplantation remained time limited, stressing the need to develop alternative sources of cells such as liver stem cells with a higher proliferation capacity for a longer-lasting effect. This time limited effect can potentially be related to death of transplanted cells with limited replicative potential.
The liver is an ideal organ for cell-based regenerative medicine, as it is also used as the recipient organ for pancreatic islets transplantation. Hepatocyte transplantation and islet transplantation are currently the two most successful techniques for demonstrating the feasibility and efficacy of cell-based regenerative medicine, beside hematopoietic progenitors and bone marrow transplantation. These are considered as first generation of a new biotechnology therapeutic approach and open the way to advanced therapies based on in vitro expanded stem or progenitor cells.
As discussed by Harding et al. in their review of PAH-/-liver repopulation in animal models by cell transplantation, an effective method of providing a selective growth advantage for the donor cells still has to be developed (5). In this context, a consensus meeting on improvement of hepatocyte transplantation success was recently published, suggesting that, in order to obtain sufficient levels of repopulation of the liver with donor cells in patients with a metabolic liver disease, some form of liver preconditioning would likely be required to enhance the engraftment and/or proliferation of donor cells. Clinical protocols for preconditioning by hepatic irradiation, portal vein embolization, and surgical resection had been developed on animal models, and clinical studies using these protocols are currently initiated in clinical practice (14, 22).
A major limitation of mature hepatocyte transplantation remains the lack of donors and the poor resistance of mature hepatocytes to cryopreservation (20). Therefore, we have for the first time in liver cell transplantation applied the domino procedure by which cells from a metabolic patient, type 1b glycogen storage disease in this particular case, are used to cure a different metabolic condition. Since the maximal cell replacement so far obtained by hepatocyte transplantation does not exceed 14% and since the recipient's own cells do not have the donor metabolic defect, there was no risk to induce a secondary glycogenolysis defect in the patient (21). This proposal from the organ procurement team, including the theoretical risk of liver adenoma, was extensively discussed with the institution's ethical review board. The donor had no adenoma, and the risk of adenoma is considered to be related to the poor metabolic equilibrium in glycogen storage disease 1 (GSD-1) patients, which would not be the case in the recipient (13, 24). Moreover, it was also considered that it would have been possible to destroy the infused cells by stopping immune suppression and inducing heterologous cell rejection. Even if we were not able to demonstrate which of the two batches of cells did participate more in the metabolic control, this concept allows for access to freshly isolated cells and by this facilitates the hepatocyte transplantation procedure.
We conclude that cell transplantation for PKU patients is an option that may correct severe hyperphenylalaninemia in poorly controlled patients being at high risk of neurological and intellectual impairment. This is a first breakthrough in the current management approach of the disease and brings new perspectives for patients and families. Provided that potency assays demonstrate that PAH activity is indeed present in candidate stem/progenitor cells, this new generation of advanced therapy medicinal products, possibly linked to preparatory regimen, can increase durability and bring these hopes to reality. Candidate cells that can replace mature hepatocytes and are close to clinical evaluation are currently available (6, 9, 12).
Footnotes
Acknowledgment
Authors declare no conflicts of interest.