
Abstract
Chondrogenesis of mesenchymal stem cells (MSCs) is typically induced when they are condensed into a single aggregate and exposed to transforming growth factor-β (TGF-β). Hypoxia, like aggregation and TGF-β delivery, may be crucial for complete chondrogenesis. However, the pellet dimensions and associated self-induced oxygen gradients of current chondrogenic methods may limit the effectiveness of in vitro differentiation and subsequent therapeutic uses. Here we describe the use of embryoid body-forming technology to produce microscopic aggregates of human bone marrow MSCs (BM-MSCs) for chondrogenesis. The use of micropellets reduces the formation of gradients within the aggregates, resulting in a more homogeneous and controlled microenvironment. These micropellet cultures (~170 cells/micropellet) as well as conventional pellet cultures (~2 × 105 cells/pellet) were chondrogenically induced under 20% and 2% oxygen environments for 14 days. Compared to conventional pellets under both environments, micropellets differentiated under 2% O2 showed significantly increased sulfated glycosaminoglycan (sGAG) production and more homogeneous distribution of proteoglycans and collagen II. Aggrecan and collagen II gene expressions were increased in pellet cultures differentiated under 2% O2 relative to 20% O2 pellets but 2% O2 micropellets showed even greater increases in these genes, as well as increased SOX9. These results suggest a more advanced stage of chondrogenesis in the micropellets accompanied by more homogeneous differentiation. Thus, we present a new method for enhancing MSC chondrogenesis that reveals a unique relationship between oxygen tension and aggregate size. The inherent advantages of chondrogenic micropellets over a single macroscopic aggregate should allow for easy integration with a variety of cartilage engineering strategies.
Keywords
Introduction
Articular cartilage has poor regenerative capacity following injury and degradation, due in part to its avascular nature. For some patients, autologous chondrocyte implantation (ACI) is a viable cartilage repair strategy; however, this procedure requires the isolation of chondrocytes via a preliminary surgery, which itself may result in further cartilage degeneration (44). Additionally, the expansion of articular chondrocytes (ACs) can result in dedifferentiation and loss of the mechanical and phenotypic properties that make the cells ideal in the first place (11, 13, 76). Adult mesenchymal stem cells (MSCs) may be an alternative autologous source for such applications due to their multipotency and relative ease of isolation and expansion (64). The use of MSCs in cartilage repair, however, will be dependent on the development of efficient and controlled chondrogenesis methods.
In vitro chondrogenesis of bone marrow-derived MSCs (BM-MSCs) in the presence of transforming growth factor-β1 (TGF-β1) was first described using high-density pellet cultures (32, 49, 82). Aggregate formation along with members of the TGF-β superfamily (49, 70) may be essential for complete in vitro chondrogenesis of MSCs. While conventional pellet culture is an effective tool for studying this process, it is not without its limitations. Typical histological analyses of these macroscopic pellets reveal heterogeneous staining of the chondrogenic-specific matrix (5, 31, 37, 49, 55, 56, 61, 70, 82). This is possibly due to fluctuating mass transport properties of the increasingly dense pellet. Minimizing transport limitations may improve homogeneity of differentiation, an essential outcome for downstream therapeutic applications.
Another critical factor in chondrogenic differentiation, and possibly in BM-MSC maintenance in general, is oxygen tension. The physiological environments of both articular cartilage and bone marrow are reported to exist within a range of 1–7% O2 (14, 36). In human ACs, expression of the essential transcription factor for chondrogenesis, (sex determining region Y)-box 9 (SOX9), is upregulated by hypoxia, resulting in increased expression of collagen II and aggrecan, the major structural components of articular cartilage (41). Additionally, hypoxia promotes the chondrocyte phenotype through SOX9-independent gene regulation (40). In recent years, a number of studies have shown the benefits of low oxygen tension with regards to BM-MSC culture and differentiation. Culture under a low oxygen environment has been shown to increase the expansion potential of BM-MSCs (20, 24, 25, 54, 86). Furthermore, posthypoxia exposure differentiation studies have shown these cells to maintain multilineage differentiation capacity with enhanced chondrogenic potential (51, 86). The few studies utilizing low oxygen during chondrogenic differentiation also indicate improved outcomes. Human adipose-derived MSCs in both alginate gels and aggregate culture under hypoxic environments showed increased chondrogenesis (36, 79). Likewise, low oxygen tension enhanced chondrogenic differentiation of high-density cultures of bovine, mouse, and rat BM-MSCs (34, 68, 69).
Recently, Ungrin et al. developed a microfabrication-based nonadhesive surface for the culture of thousands of individual aggregates of embryonic stem cells to improve embryoid body homogeneity and differentiation (78). With a commercial tissue culture product employing this microwell surface now available, we evaluated the effectiveness of such a technology for enhancing chondrogenesis of human BM-MSCs. Due to the promising indications but lack of clarity regarding the role of oxygen tension during chondrogenesis of human BM-MSCs, the oxygen environment was varied between normoxic (20% O2) and hypoxic (2% O2) for this new culture system as well as for conventional pellet culture. We show here that a long-term low oxygen environment during chondrogenic induction has beneficial effects on the differentiation of human BM-MSCs in a conventional pellet culture. Furthermore, creating smaller cell aggregates under low oxygen tension resulted in substantial increases in the changes characteristic of chondrogenic differentiation compared to conventional pellet cultures in both environments. Specifically, we show that chondrogenic induction of BM-MSC micropellets formed in AggreWell™ plates under low oxygen tension results in considerably increased sulfated glycosaminoglycan (sGAG) production, uniform distribution of matrix components, and enhanced expression of genes associated with BM-MSC chondrogenesis. Thus, we have developed a new method for enhanced chondrogenic differentiation of BM-MSCs that possesses properties ideal for incorporation with current platforms for cartilage repair.
Materials and Methods
Human Bone Marrow-Derived Mesenchymal Stem Cell Isolation and Culture
Full informed patient consent was obtained in all cases and ethical approval granted through the Mater Health Services Human Research Ethics Committee in accordance with the Australian National Health and Medical Research Council's Statement on Ethical Conduct in Research Involving Humans. Approximately 10 ml bone marrow was taken from iliac crest of healthy donors. The sample was diluted 1:1 with phosphate-buffered saline (PBS) and underlayed with 12 ml Ficoll-Paque Plus (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Tubes were spun at 535 × g for 20 min. Interface cells were washed and resuspended in low-glucose Dulbecco's modified Eagle's medium (DMEM-LG; Gibco Life Technologies, Grand Island, NY) supplemented with 20% fetal bovine serum (FBS; Gibco) and 50 μg/ml gentamicin (Amersham Pharmacia Biotech, Uppsala, Sweden) and placed in tissue culture flasks. After 48 h, nonadherent cells were removed by washing with PBS and remaining adherent cells further cultured with medium changes every 3–4 days. Cells generally approached confluence after 14–20 days and were then passaged and expanded. After the second passage, cells were immunophenotyped by flow cytometry (monoclonal antibodies from BD Biosciences Pharmingen, San Diego, CA) and were functionally assessed for differentiation potential. Cells were deemed MSC if they were CD45–, CD73+, CD90+, CD105+, and showed adipogenic, osteogenic, and chondrogenic differentiation potential as described previously (8).
For these experiments, second passage BM-MSCs were expanded in DMEM-LG supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (1% PS; Gibco) in an incubator with a 2% O2 atmosphere due to the aforementioned evidence of the benefits of hypoxic preconditioning on MSC chondrogenesis. BM-MSCs from three different donors at fourth passage were used in chondrogenic assays.
Chondrogenic Differentiation
BM-MSCs were differentiated as conventional pellet cultures in 15-ml polypropylene tubes or micropellet cultures formed in AggreWell™ 400 plates (STEMCELL Technologies, Vancouver, BC, Canada). BM-MSCs were grown to near confluence, detached using recombinant trypsin replacement (TrypLE; Gibco), and placed in serum-free chondrogenic induction medium consisting of high-glucose DMEM (DMEM-HG; Gibco) containing 10 ng/ml TGF-β1 (PeproTech, Rocky Hill, NJ), 10–7 M dexamethasone (Sigma, St. Louis, MO), 200 μM ascorbic acid 2-phosphate (Sigma), 100 μg/ml sodium pyruvate (Sigma), 40 μg/ml proline (Sigma), 1× ITS+ (Gibco), and 1% PS. Pellet or micropellet cultures were formed by centrifuging 2 × 105 cells at 500 × g in chondrogenic induction medium and then culturing in a 2% O2 or 20% O2 atmosphere for a further 14 days. In this study, we used TGF-β1, which is known to induce chondrogenic differentiation of MSCs but at a lesser rate than TGF-β3 (5). This allowed us to evaluate micropellet differentiation at a time point (14 days) where chondrogenesis would be initiated in conventional pellet cultures but still at an early stage (63).
Sulfated Glycosaminoglycan Quantification
Medium from pellet and micropellet cultures was collected and stored at −80°C at each medium replacement, every 3–4 days. At the end of 14 days, micropellets were centrifuged to a single pellet. Micropellets and pellets were digested with 1.6 U/ml papain (Sigma) at 60°C overnight. The sGAG content and DNA were quantified with 1,9-dimethymethylene blue (DMB; Sigma) and Hoechst 33342 (Molecular Probes, Eugene, OR) as described in detail by Liebman and Goldberg (47). Shark chondroitin sulfate (Sigma) and calf thymus DNA (Sigma) were used as the respective standards. Additionally, the DMB assay was used to quantify sGAGs released into the medium collected at days 3, 6, 10, and 14.
Histology and Immunohistochemistry
At the end of 14 days, micropellets and pellets were fixed in 4% formaldehyde, embedded in Tissue-Tek OCT compound (Sakura Finetek, Tokyo, Japan), and snap-frozen in liquid nitrogen. Samples were cryosectioned and stored at −80°C until use. Before staining, the sections were rinsed in 70% ethanol and Tris-buffered saline (TBS) to remove OCT compound. To detect proteoglycan deposition sections were stained with 0.1% toluidine blue (ProSciTech, Thuringowa, QLD, Australia) in 1% NaCl solution (pH 2.3). Organization of fibrillar collagen was detected by staining with 0.1% Picrosirius red F3B (ProSciTech) as previously described (12) and visualizing with an Olympus BX61 microscope equipped with polarizing filters.
Localizations of collagen I and collagen II were determined by double immunofluorescence staining (IF). Briefly, the sections were digested with 0.01% pepsin (Sigma) in 0.01 M HCl (pH 2) at 37°C for 10 min, followed by 0.1% hyaluronidase (Sigma) in PBS (pH 5) at room temperature (RT) for 30 min. Cells were permeabilized with 0.1% Triton X-100 for 5 min and blocked for 30 min at RT in TBS containing 2% bovine serum albumin and 2% normal goat serum. The sections were then stained with a 1:50 dilution of both polyclonal rabbit anti-collagen I (Cedarlane Labs, Burlington, ON, Canada) and monoclonal mouse anti-collagen II (Lab Vision, Fremont, CA) primary antibodies for 2 h at RT. This was followed by incubation with a mixture of secondary antibodies containing Alexa Fluor 568-conjugated goat anti-rabbit IgG and Alexa Fluor 488-conjugated goat anti-mouse IgG (1:200 dilution; both from Molecular Probes) for 1 h at RT. After each staining step, unbound antibodies were washed with TBS containing 0.2% Tween-20. Nuclei were counterstained with Hoechst 33342 for 5 min at RT. The sections were mounted and examined with an Olympus BX61 fluorescence microscope. Negative controls without primary antibodies were used for background correction.
Relative Gene Expression Analysis
On day 14, RNA was collected from micropellets and mechanically disrupted conventional pellets using the RNEasy Mini Kit (Qiagen, Valencia, CA) as per the manufacturer's instructions. RNA was also collected from day 0 monolayer BM-MSCs. RNA samples were treated with DNase I (0.1 U/μl final; Fermentas, Glen Burnie, MD) for 30 min at 37°C and then heat inactivated at 65°C for 5 min in the presence of 2.5 mM EDTA. DNase I-treated RNA samples (50 ng) were reverse transcribed using SuperScript III RT and oligo(dT)20 in the presence of RNaseOUT (all from Invitrogen) as per the manufacturer's instructions and stored at −80°C until analysis.
Real-time quantitative polymerase chain reaction (qPCR) was performed using a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) and Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen). The cycling parameters were 50°C for 2 min, 95°C for 2 min, and then 95°C for 3 s and 60°C for 30 s for a total of 40 cycles. The primers used are shown in Table 1 and were all from previously published papers (52, 72, 77, 81). Results were analyzed using the 2–ΔΔCt method relative to the housekeeping gene cyclophilin A due to the instability of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in oxygen-dependent studies (7, 21, 85). Specificity of products was confirmed by melt curve analysis and 3% agarose gel electrophoresis.
Primers Used for Real-Time Quantitative Polymerase Chain Reaction

GAPDH, glyceraldehyde-3-phosphate dehydrogenase; SOX9, (sex determining region Y)-box 9; Runx2/Cbfa1, runt-related transcription factor 2/core-binding factor α1.
Cyclophilin A was stable among day 14 differentiated BM-MSCs, but differed in monolayer BM-MSCs and thus could not be used to compare between days 0 and 14. GAPDH, however, was stable between 2% O2 micropellets and monolayer BM-MSCs. Thus, for some genes, we quantified the change in 2% O2 micropellets compared to monolayer BM-MSCs and used this in conjunction with the change among conditions calculated using cyclophilin A to indirectly estimate the change in the conventional pellets from monolayer BM-MSCs. However, this was deemed to be more susceptible to error and was only used as an indication of the general magnitude and direction of changes in pellets compared to undifferentiated BM-MSCs. Therefore, all data shown and subjected to statistical analysis are compared to the current standard for BM-MSC chondrogenesis, 20% O2 pellets, and with cyclophilin A as a reference gene.
Statistical Analysis
SPSS 17.0 (SPSS Inc., Chicago, IL) was used for one-way analysis of variance (ANOVA) with Tukey post hoc tests to assess statistical significance, which was defined as p < 0.05. For qPCR data, statistical analysis was conducted on the ΔCt values and the mean fold increase and 95% confidence intervals are represented by 2–ΔΔCt evaluated at the mean ΔΔCt, at the lower confidence limit of ΔΔCt, and at the upper confidence limit of ΔΔCt between conditions (83).
Results
Human BM-MSC Micropellet Development in AggreWell™ Plates
The feasibility of creating micropellets of BM-MSCs in the AggreWell™ plates was evaluated by varying the number of cells per well from 2 × 105 to 1 × 104 (results not shown). We found that consistent aggregates could be formed under both 20% and 2% O2 using 2 × 105 cells/well, a common number of MSCs used in conventional pellet culture. At this density, aggregates formed under both oxygen environments within 14 days, albeit with different morphologies (Fig. 1). Micropellets under 2% O2 appeared to be more loosely aggregated while those under 20% O2 formed smaller compacted masses. Micropellets from both oxygen environments were easily collected for analysis at day 14 using only a pipette to dislodge them from microwells. All subsequent studies therefore used 2 × 105 cells per centrifuge tube or per well (~170 cells/microwell).

Morphological changes of human BM-MSC micropellets over 14 days of chondrogenic induction in AggreWell™ plates under 20% and 2% O2 environments. Scale bar: 200 μm.
Proteoglycan Production in Normoxic and Hypoxic Micropellets and Pellets
The proteoglycan production of micropellets was compared to that of conventional pellets by quantifying the total amount of sGAGs released over the course of chondrogenic induction and the amount retained within the aggregates' matrices. While 20% O2 pellets displayed a relatively level profile, the amount released by 2% O2 pellets showed a slightly increasing profile over 14 days (Fig. 2A). Micropellets differentiated at 20% O2 generally displayed a steady but elevated release profile of sGAGs, while 2% O2 micropellets had a distinctly linear sGAG release profile (Fig. 2B). The 2% O2 pellets retained nearly twice the fraction of their total sGAGs as the 20% O2 pellets (Fig. 2C) and correspondingly produced twice as much per microgram of DNA within the matrix (Fig. 2D). Micropellets cultured at 2% O2, meanwhile, produced 8.2- and 4.0-fold more total sGAGs than pellets at 20% and 2% O2, respectively (Fig. 2C). The increase was also reflected in the amount of sGAGs retained with 6.3- and 1.6-fold more sGAGs within the aggregate mass of 2% O2 micropellets. While 2% O2 micropellets retained less than half the fraction of their total as 2% O2 pellets (Fig. 2C), the increase in total released was of such a magnitude that the amount within the aggregates' matrices per DNA was not significantly different (Fig. 2D). The compact 20% O2 micropellets were typically found to have very little DNA after 14 days and a low amount of sGAGs within the collected aggregate mass. We suspected that these micropellets experienced substantial cell death as there were often relatively few remaining after 10–14 days. Per DNA, the amount of sGAGs was statistically equivalent to that produced in the conventional 20% O2 pellet (Fig. 2D).

Production of sGAGs by human BM-MSC pellets and micropellets differentiated under 20% and 2% O2. The release profile of sGAGs over 14 days was determined by the quantity released into the supernatant between medium exchanges for (A) conventional pellet cultures and (B) micropellet cultures. (C) The total amount of sGAGs produced over 14 days and the amount retained within the pellets and micropellets after 14 days. (D) The amount of sGAGs that was retained within the pellets and micropellets normalized to DNA. All values are mean ± SD for n = 10 samples from three independent experiments. Statistical significance was determined by ANOVA with Tukey post hoc tests. *p < 0.05 compared to 20% O2 pellets; †p < 0.05 compared to 2% O2 pellets. sGAGs, sulfated glycosaminoglycans.
Matrix Distribution in Hypoxic Micropellets and Normoxic and Hypoxic Pellets
After 14 days the distributions of proteoglycans and collagens in the pellets and 2% O2 micropellets were visualized using toluidine blue and polarized light imaging of picrosirius red, respectively. Due to the small size of remaining 20% O2 micropellets, we did not cryo-section samples for this evaluation. In pellets, staining of proteoglycans was heterogeneous with the darkest staining around the periphery while 2% O2 micropellets exhibited homogeneous staining (Fig. 3A). Throughout 2% O2 micropellets the cells appeared as round, chondrocyte-like cells embedded within lacunae. The distribution of collagen fibers was similar in the 20% O2 pellets and 2% O2 pellets, with thicker fibers aligned along the periphery and random thin fibers in the central region (Fig. 3B, C). A random meshwork of fibers with no regional organization was observed surrounding the cells in the 2% O2 micropellets. The collagen matrix surrounding the differentiating cells was also assessed by visualizing collagen I and collagen II using IF. Collagen I was detected throughout pellets differentiated under both oxygen environments and consistent with toluidine blue staining, collagen II was only detected in areas along the periphery (Fig. 4A–C). In contrast, positive immunohistochemical stainings of both collagen I and collagen II were visualized essentially throughout the matrix of 2% O2 micropellets, although collagen I was generally more intense in the inner regions.
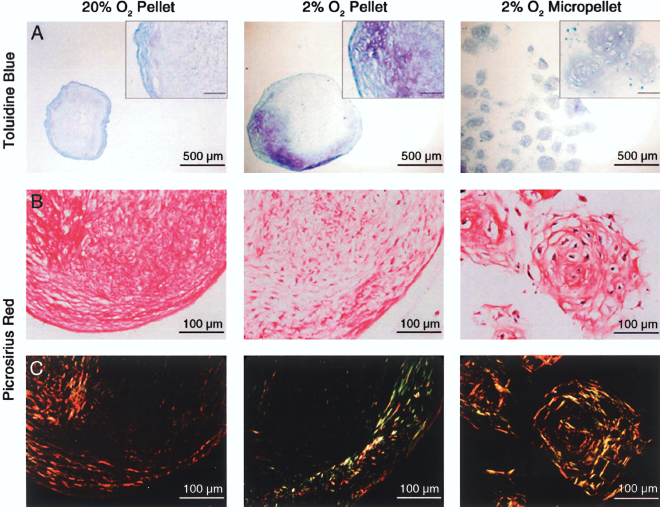
Distribution of proteoglycans and organization of collagen fibers in 20% O2 pellet, 2% O2 pellet, and 2% O2 micropellet cultures of human BM-MSCs in chondrogenic medium for 14 days. Fixed cryosections were stained with (A) toluidine blue for proteoglycans or (B) picrosirius red for collagen and viewed under normal bright field. (C) Birefringent collagen fibers of the picrosirius red-stained sections were visualized with polarized light microscopy. Inset scale bars: 100 μm.

Localization of collagen in 20% O2 pellet, 2% O2 pellet, and 2% O2 micropellet cultures of human BM-MSCs maintained in chondrogenic medium for 14 days. Fixed cryosections were double-stained with (A) anti-collagen I and (B) anti-collagen II antibodies, as described in Materials and Methods, with Hoechst 33342 as a counterstain for nuclei. Positive immunolocalization of collagen appears as red (collagen I) or green (collagen II) in the extracellular matrix, and nuclei are blue. Negative controls where the primary antibodies were omitted (images not shown) were used for background correction. (C) Composite images of (A) and (B) were digitally processed using ImageJ software. The overlapping regions (yellow) indicate colocalization of collagens I and II. Inset scale bars: 1 mm.
Gene Expression of Normoxic and Hypoxic Micropellets and Pellets
The extent of chondrogenesis was distinguished by quantifying the relative gene expression of collagen II, aggrecan, and SOX9. We also evaluated a variety of genes more prevalent in MSCs, fibrocartilage, hypertrophic chondrocytes, and osteoblasts: collagen I, versican, collagen X, runt-related transcription factor 2/core-binding factor α1 (Runx2/Cbfa1), and osteocalcin.
After 14 days of chondrogenic induction, qPCR analysis revealed no significant difference in SOX9 expression between 2% O2 and 20% O2 pellets (Fig. 5A). SOX9 in 2% O2 micropellets, meanwhile, showed an average increase of 7.5-fold over 20% O2 pellets and 3.0-fold over 2% O2 pellets. Aggrecan was increased 12- and 350-fold in 2% O2 pellets and 2% O2 micropellets, respectively, compared to 20% O2 pellets (Fig. 5B). Collagen II was increased on average 1,500-fold in 2% O2 pellets and 33,000-fold in 2% O2 micropellets compared to 20% O2 pellets (Fig. 5C). We screened 20% O2 micropellet samples for aggrecan and collagen II gene expression and found these to be expressed at similar levels as in 20% O2 pellets (data not shown) and taking into consideration the sGAG results, did not continue with the full panel of genes for 20% O2 micropellets.

Fold-change in mRNA levels of common markers for chondrogenesis, hypertrophy, and osteogenesis in human BM-MSC pellets and micropellets differentiated under 2% O2 compared to conventional 20% O2 pellets. After 14 days in chondrogenic culture the relative gene expression levels of (A) the transcription factors SOX9 and Runx2/Cbfa1, (B) the proteoglycans aggrecan and versican, (C) collagens I, II, and X, and (D) the bone-forming marker osteocalcin were analyzed by real-time quantitative polymerase chain reaction. All values are the mean fold-change relative to 20% O2 pellets normalized to cyclophilin A for n = 7 samples from three independent experiments. Error bars represent 95% confidence intervals. Statistical significance was determined by ANOVA with Tukey post hoc tests. *p < 0.05 compared to 20% O2 pellets; †p < 0.05 compared to 2% O2 pellets. SOX9, (sex determining region Y)-box 9; Runx2/Cbfa1, runt-related transcription factor 2/core-binding factor α1.
Collagen I was not significantly different between pellet cultures but increased 4.2-fold in 2% O2 micropellets compared to 20% O2 pellets (Fig. 5C). Comparing 2% O2 micropellets to monolayer MSCs using GAPDH revealed a 2.2-fold increase (data not shown), meaning that the monolayer MSCs expressed collagen I at an intermediate level between pellets and 2% O2 micropellets. Versican, on the other hand, was not significantly different among the differentiated conditions (Fig. 5B). Collagen X followed the trend of collagen II, although it was highly variable among samples even within the same donor. Still, there was a significant 21-fold increase in collagen X in 2% O2 pellets and 1,600-fold in 2% O2 micropellets compared to 20% O2 pellets (Fig. 5C). Despite this trend, there was no significant difference in Runx2/Cbfa1 or osteocalcin gene expression in 2% O2 micropellets compared to 20% O2 pellets (Fig. 5A, D), although Runx2/Cbfa1 was significantly lower in 2% O2 pellets compared to micropellets.
Discussion
In this study, we investigated chondrogenesis of human BM-MSCs in conventional pellet culture compared to micropellets and the effects of oxygen tension in these different aggregates. The importance of MSC aggregation has been partially explained by the role of Jagged-1-mediated Notch signaling in the chondrogenesis pathway (29). Notch signaling, which is activated by cell–cell contact, must be initiated for MSCs to begin chondrogenesis but must later be switched off for differentiation to continue (61). TGF-β3 was recently discovered to downregulate Notch gene expression and protein levels in MSCs, resulting in the SOX9-mediated upregulation of collagen II (26). Thus, TGF-β may be a key regulator resulting in the transience of Notch signaling that is critical for chondrogenesis of MSCs. Given that hypoxia reportedly enhances Notch signaling (27), oxygen tension may also be a crucial factor. We hypothesize that the benefits of low oxygen on in vitro MSC chondrogenesis may be abrogated somewhat by the limitations of current chondrogenic protocols. Specifically, we believe that the mass transport properties and self-induced oxygen gradient of macroscopic pellet culture are substantial obstacles to robust chondrogenesis.
The most critical known soluble factors for chondrogenic differentiation of MSCs range in molecular weight from 0.39 (dexamethasone) to 25 kDa (TGF-β). Mass transport studies of tissue-engineered cartilage constructs have shown a relationship between increasing tissue density and decreasing diffusivity, even for molecules as small as glucose (0.18 kDa) (10, 22, 42). In pellet culture, the microarchitecture has been observed to resemble that of articular cartilage, with collagen fibers aligned parallel to the surface at the surface and perpendicular to the surface in the deeper layers (6, 31, 56). Interestingly, in articular cartilage, Leddy and Guilak discovered that small and large molecular weight dextrans (3 and 500 kDa) had the greatest diffusivity through the surface zone, while midrange dextran (40 and 70 kDa) had lower diffusivity through the surface zone (43). Over the course of differentiation of pellet-cultured MSCs, it is likely that matrix accumulation results in the diffusivity approaching that of articular cartilage. Therefore, strategies that minimize the mass transport limitations in MSC chondrogenesis have the potential to maximize the efficiency and homogeneity of differentiation. The wells of AggreWell™ plates each consist of approximately 1,200 microwells and allow for the creation of very small diffusional distances in adjacent but physically separate microscopic aggregates. Using the assumption that cells form perfect spheres of an equal initial total volume as a single perfect sphere of cells, distributing the cells across the AggreWells™ would decrease the radius of aggregates by approximately 11 times. This would result in an approximate 11-fold increase in the surface area-to-volume ratio, which would substantially enhance mass transport.
Over 14 days of chondrogenic induction, BM-MSC micropellets cultured in AggreWell™ plates under a 2% O2 environment demonstrated substantially increased chondrogenesis over conventional pellet cultures differentiated under both 2% and 20% O2. While the endpoint for analysis was at 14 days, we monitored development over the course of chondrogenic induction morphologically and by quantifying the release of sGAGs. The accumulation of sGAGs begins in the early stages of MSC chondrogenesis (5) and the quantification of these proteoglycan modifications can give an indication of aggrecan production. Aggrecan is largely responsible for providing the compressive stiffness of articular cartilage (65) and obtaining cartilaginous tissues in vitro with similar aggrecan composition remains a challenge (38, 56). In accordance with results seen in pellets of normal ACs (29), our BM-MSC pellets under 2% O2 retained more of their total sGAGs within the cell mass and correspondingly produced more per DNA than the 20% O2 pellets. These 2% O2 pellets were also noticeably larger when viewed with the naked eye, a result others have reported for low oxygen-preconditioned and low oxygen-exposed pellet cultures of MSCs and ACs, which is likely primarily a product of increased proteoglycan content (29, 51, 53, 86). The release profiles for 20% O2 micropellets were generally elevated over pellet cultures but per DNA within the cell mass, they performed no better than conventional 20% O2 pellets. The difference in sGAG production over 14 days in 2% O2 micropellets, on the other hand, was striking. These micropellets swelled in size like their macroscopic counterparts under 2% O2 and had a highly linear increase in sGAG release over the entire 14 days. Although 2% O2 micropellets retained a lower fraction of their total than 2% O2 pellets, the total sGAG production was so significantly elevated that the quantity of sGAGs per DNA within the mass was similar.
We histologically assessed the distributions of proteoglycans as well as collagen in the 20% O2 pellets, 2% O2 pellets, and 2% O2 micropellets. In conventional pellets collagen II and proteoglycans are typically observed either in the center (5, 49, 61, 82) or at the periphery (5, 31, 37, 55, 56, 70). Although, due to multiple variables between these studies, it is difficult to discern the reasoning for the different patterns observed, the fluctuating mass transport properties of the differentiating pellet may play a role. In larger pellets, central necrosis has even been observed (17). In this study, both 20% and 2% O2 pellets displayed mostly peripheral staining of proteoglycans and collagen II but showed collagen I throughout. Although micropellets were not a perfectly uniform size the distribution of proteoglycans and collagens appeared homogeneous. The micropellets also displayed random orientation of collagen fibers similar to the middle to deep zones of normal articular cartilage (19, 56, 65). There were no structurally aligned fibers around the periphery of the micropellets as was seen in conventional pellets and as has been observed by others (56). Furthermore, while collagen I was variably distributed in the micropellets, collagen II appeared throughout micropellets after only 14 days with TGF-β1.
Analysis of cartilage-specific genes using qPCR revealed collagen II and aggrecan to be significantly upregulated in 2% O2 pellets but further increased in 2% O2 micropellets along with SOX9. Collagen II in 2% O2 micropellets was elevated 33,000-fold over 20% O2 pellets and approximately 785,000-fold more than undifferentiated BM-MSCs (data not shown). In 2% O2 micropellets with 10 ng/ml TGF-β1 we measured even greater increases in collagen II expression at 14 days than has been reported for normoxic pellet cultures supplemented with 100 ng/ml of the more potent TGF-β3 (61). Due to the extremely high level of sGAGs produced in 2% O2 micropellets compared to pellet cultures, in addition to aggrecan we also evaluated versican expression, which is high in dedifferentiated ACs but low in normal articular cartilage (4, 52). Versican is also expressed in undifferentiated BM-MSCs (75) and an increase in its gene expression has been described as an early event in pellet culture chondrogenesis (5). In this study, unlike aggrecan expression which increased on average 350-fold in 2% O2 micropellets compared to 20% O2 pellets, there was no significant difference in versican expression among the different conditions.
The inability to maintain high levels of collagen II expression relative to collagen I in primary ACs expanded in vitro potentially reduces their efficacy in ACI procedures (4, 67). While collagen I is highly expressed in BM-MSCs (75) and extremely low in normal ACs (4, 50), its average expression during pellet culture chondrogenic induction of BM-MSCs has been observed by some to increase along with collagen II and collagen X over the long term (35, 55, 63). We found that collagen I was significantly higher in 2% O2 micropellets compared to 20% and 2% O2 pellet cultures, both of which had decreased levels compared to monolayer BM-MSCs. However, we think that with continued induction beyond 14 days the pellets would continue to increase their average collagen II and collagen X expression as well as their average collagen I expression.
Perhaps the principal challenge of using BM-MSCs for cartilage repair will be the ability to obtain a stable chondrogenic phenotype. In this study, we evaluated the hypertrophic and osteogenic markers collagen X, Runx2/Cbfa1, and osteocalcin. Changes in collagen X expression among conditions mirrored the changes seen with collagen II, as has been reported previously with conventional pellets (5, 56, 59, 63, 71, 86). This could be a significant obstacle with BM-MSCs, as Pelttari et al. showed that ectopic implantation of in vitro differentiated BM-MSC pellets in SCID mice led to calcification and vascular invasion at the implant site, whereas no such problems were observed with chondrocyte-derived pellets (63). However, we believe that our improved methodology will allow for better control when investigating methods to reduce collagen X expression and, likewise, collagen I expression.
Collagen X expression in hypertrophic chondrocytes is regulated by Runx2/Cbfa1 (18, 30, 84), a critical transcription factor for osteoblast differentiation and bone formation (16, 39, 62) and an important regulator of human BM-MSC osteogenic differentiation (1, 28, 48, 73). Despite greatly increased levels of collagen X, there was no significant difference in Runx2/Cbfa1 gene expression compared to 20% O2 pellets. However, while increased Runx2/Cbfa1 activity is critical for osteogenesis of human BM-MSCs, its mRNA expression may not change even during osteogenic differentiaton (73). We therefore examined expression of another Runx2/Cbfa1 target, osteocalcin, as it is regulated during osteogenesis and has been indicated as a hypertrophic marker in chondrocytes and in human BM-MSC pellet cultures (3, 23, 46, 55, 74). Despite the apparent advanced stage of the 2% O2 micropellets, there was no significant difference in osteocalcin expression among conditions. While 14 days may be too early to see such changes even in the micropellets, in addition to observing reduced osteogenesis of BM-MSCs under prolonged low oxygen tension (20), others have noted that even temporary hypoxic exposure inhibits BM-MSC osteogenic differentiation (66). Consequently, we believe that with further optimization micropellets under low oxygen tension may offer a means to reach a stable chondrogenic phenotype.
Here we showed the benefit of low oxygen tension during chondrogenic induction of human BM-MSC using both conventional pellet culture and a new micropellet culture. The benefits of low oxygen tension appear to be maximized by creating very small aggregates that limit mass transport and oxygen diffusion gradients. While the undesired collagen X was greatly upregulated in 2% O2 micropellets, this problem persists in conventional BM-MSC pellet culture as well. A plasma polymer coating has recently been reported to decrease collagen X (58) and such modifications would be easily applicable to microfabricated surfaces such as the inserts of AggreWells™. Soluble factors such as chondroitin sulfate (9) and parathyroid hormone-related protein (33, 37), however, will likely be the most commonly investigated solutions and, as shown here, a micropellet system may maximize the efficiency of such strategies. Additionally, we have developed a membrane bioreactor that is able to aid conventional pellet cultures in retaining sGAGs within their matrices (15) and believe micropellet cultures will be easily integrated with such technology. With a view towards future tissue engineering, we believe the benefits of performing MSC chondrogenesis in this way is to date unparalleled. Homogeneous differentiation of the cell population can be achieved more rapidly and the physical nature of the chondrogenic micropellets should make them easy to integrate with a variety of stem cell-based cartilage and meniscal repair technologies such as intra-articular injections (45, 57), hydrogels (60), and scaffolds (2, 80).
Footnotes
Acknowledgments
This work is supported by The University of Queensland Early Career Researcher Grant Scheme and in part by the Australian Research Council Discovery Grants Scheme. The Whitaker Foundation provided support for B.D.M. with a Whitaker International Fellows Award (2008-09). The authors thank Paul Addison for technical support with histology, Professor Julie Campbell for supporting this project with shared equipment and lab space, and the Australian Stem Cell Centre for generously allowing access to analysis equipment. B.D.M. also thanks his Ph.D. advisor, Dr. Monica Hinds of Oregon Health & Science University, for her selfless support of his fellowship.