
Abstract
Cell transplantation has emerged as a novel therapeutic strategy for periodontitis, and the adoption of cell pellet offers advantages by secreting abundant extracellular matrix (ECM) and eliminating the adverse effect of cell carriers. This study aimed to fabricate scaffold-free periodontal ligament stem cell (PDLSC) pellets (MUCPs) and to evaluate their regeneration potential. We constructed monolayer cell pellets (MCPs) by fabricating and culturing multilayered cell sheets (MUCS) and constructed MUCPs from the MUCS. Immunochemistry, scanning electron microscope, real-time PCR, and Western blot analysis showed higher levels of COL-I, COL-III, fibronectin, and laminin in the MUCPs. Furthermore, the massive increase in ECM secretion improved cell adhesion, migration, and proliferation. Finally, upon transplantation into the omentum sac and periodon-tal defects, all the transplants formed regular aligned cementum/PDL-like complex, but the mineral deposit and fiber alignment were more obvious in the MUCPs than in the MCPs. Altogether, our results suggest that MUCPs may be a promising alternative to periodontal repair for future clinical application.
Keywords
Introduction
Cell sheet engineering has pioneered the development of new therapeutic approaches for periodontal defects and shown efficacy in periodontal regeneration (15,16,22). However, conventional cell sheet engineering is confronted with two main obstacles: first, the biodegradable scaffolds incorporated with the cell sheet used for the mechanical support could cause immunogenicity, disease transmission, or inflammatory reaction upon implantation (6,7); second, the feeble biomechanical strength of the cell sheet cannot fulfill the clinical demands (15,21).
The extracellular matrix (ECM) is a complicated network composed of several structural proteins including fibronectin, laminins, collagens, and functional biological scaffold materials (1,19). The ECM provides a wide range of biochemical and mechanical cues to the cells and acts as a reservoir for many signal molecules that are critical in modulating various cellular functions such as migration, growth, and differentiation (24,36). Monolayer cell sheets, fabricated using a thermoresponsive system, contain less ECM and without proper support cannot retain its configuration and stiffness, preventing its use in three-dimensional (3D) large restoration areas (1,15).
ECM secretion involves a dynamic equilibrium with its surrounding microenvironment, and changes in ECM microstructure and components may lead to the alterations in mechanical strength (29). Our previous studies found that in vivo transplantation of the cell pellets could form a regular aligned cementum/periodontal ligament (PDL)-like complex (40,41). However, some fractures were observed within the periodontal ligament stem cell (PDLSC) pellets after 10 days of cultivation, which is thought to be a sign of inadequate nutrition in the area (40).
The preferential anchorage of the mesenchymal cells derived from the periodontal ligament over epithelial cells on the diseased root is the key to successful reattachment of the periodontal ligament. Unfortunately, PDLSCs have been shown to proliferate and anchor on the root surface at a lower rate than epithelial cells, leading to the formation of long, junctional epithelium. This is partly attributed to the lower adhesive and proliferative capabilities of PDLSCs. Fibronectin, laminin, and collagen-I (COL-I) have been demonstrated to regulate the composition and stability of the ECM (1,37). Therefore, we hypothesize that PDLSCs can settle on and cover the diseased root with greater ease in an ECM-rich environment. In this study, we modified the fabrication by using multilayered cell pellets (MUCPs) as a substitute for the previously reported monolayered cell sheet (MCS) with the intent to reduce the unfavorable effect of the condensed ECM on the cell vitality in the center of the MCS.
Materials and Methods
Animals
Sprague–Dawley (SD) adult female rats obtained from Harlan (3 months old; Xi'an, China) were used for PDLSC culture and as host rats. All animal experiments were conducted in accordance with the committee guidelines of the Fourth Military Medical University for animal experiments, which met the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals. SD female rats weighing 150–200 g were obtained from the Laboratory Animal Research Center at the Fourth Military Medical University (Xi'an, China) and maintained in a 12-h light/12-h dark, temperature-controlled room with Purina rodent chow and sterile water available ad libitum.
Preparation of Treated Dentin Matrix (TDM)
The TDMs were prepared as previously described (20). Briefly, the extracted molars of SD adult rats were harvested, and the periodontal ligament tissues and cemen-tum were carefully scraped away from the roots. Inner dental pulp tissue, predentin, and partial root dentins were removed by grinding into dentin matrixes (DMs). DMs were exposed to 17% ethylenediaminetetraacetic acid (EDTA; Sigma-Aldrich, St. Louis, MO, USA) for 3–4 min, washed with deionized water for 5 min, exposed to 5% EDTA for 2 min, and washed in deionized water for 5 min. DMs were maintained in sterile phosphate-buffered saline (PBS; Gibco BRL, Gaithersburg, MD, USA) with 100 U/ml penicillin (Gibco BRL) and 100 mg/ ml streptomycin (Gibco BRL) for 72 h, after which time they were washed in sterilized deionized water for 5 min and finally stored in conventional culture media [minimum essential medium with α-modification (α-MEM); Hyclone, Logan, UT, USA] with 50 U/ml penicillin and 50 mg/ml streptomycin at 4°C (20).
PDLSC Isolation
Rat periodontal fibroblasts were isolated and cultured following the procedures described previously (35). The periodontal ligament from the middle third of the extracted molar root was gently scraped from the surface of the root, cut into small pieces under the microscope, and cultured with complete Dulbecco's modified Eagle's medium (DMEM; Gibco BRL) supplemented with 10% fetal bovine serum (FBS; Hyclone), 0.292 mg/ ml glutamine (Invitrogen, Carlsbad, CA, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco BRL) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The explants were maintained in six-well culture plates (Costar, Cambridge, MA, USA) for 2 weeks until proliferating fibroblasts were subconfluent. To isolate putative stem cells, primary cultures of periodontal ligament cells (1 × 104 cells) were seeded into 90-mm culture dishes (Costar) and cloned as previously reported (34).
Colony-Forming Efficiency
A total of 1 × 103 third passage PDLSCs were seeded into a 75-cm2 cell culture flask (Costar) and cultured in the above medium. The medium was changed every 2 days. After incubating for 7 days, PDLSCs were observed and photographed under a phase-contrast inverted microscope (Olympus Optical, Tokyo, Japan). Aggregates of >50 cells were scored as colonies. The experiment was repeated at least three times.
Adipogenic Differentiation
A total of 1 × 105 third passage PDLSCs were seeded into a six-well plate. When PDLSCs reached 80% confluence, the medium was changed to the complete DMEM supplemented with 10% newborn bovine serum (NBS; Hyclone), 2 mM insulin (Sigma), 0.5 mM isobutylmeth-ylxanthine (IBMX; Sigma), and 10 nM dexamethasone (Sigma) for 14 days. The cells were then washed three times in PBS, fixed in 4% paraformaldehyde (Keshi, Chengdu, China) for 10 min, and incubated in 0.3% Oil Red O (Sigma) solution for 15 min. Following this, the plates were washed with PBS three times and were then routinely observed and photographed under a phase-contrast inverted microscope.
Osteogenic Differentiation
A total of 1 × 105 PDLSCs were seeded and cultured in a six-well plate until 80% confluence. Then the culture medium was changed to osteogenic-inducing medium containing 10% NBS, 5 mM L-glycerophos-phate (Sigma), 100 nM dexamethasone, and 50 mg/ml ascorbic acid (Sigma). The medium was changed every 2 days for 14 days (13,26,27). The cells were washed twice with PBS, subsequently fixed in 4% paraformal-dehyde for 10 min, and then incubated in 0.1% alizarin red solution (Sigma) in Tris–HCl (pH 8.3; Bio-Rad, Hercules, CA, USA) at 37°C for 30 min. After washing twice with PBS, cells were observed and photographed using a microscope.
Immunocytochemistry (ICH)
PDLSCs were digested and seeded onto 0.8 cm × 0.8 cm coverslips (Ciglas, Haimen, China) in a 24-well plate (CoStar), cultured for 1 day, and stained. PDLSCs were fixed with 4% paraformaldehyde for 10 min, washed three times with PBS, and then incubated for 1 h with the following primary antibodies: (i) polyclonal rabbit anti-COL-I at a 1:100 dilution, (ii) polyclonal rabbit anti-fibronectin at a 1:100 dilution, (iii) polyclonal rabbit anti-laminin at a 1:100 dilution, (iv) monoclonal mouse anti-osteocal-cin (OCN) at a 1:50 dilution, (v) polyclonal rabbit anti-alkaline phosphatase (ALP) at a 1:100 dilution, and (vi) monoclonal mouse anti-osteonectin (ON) at a 1:50 dilution. The primary antibodies were purchased from Abcam Biotechnology (Cambridge, UK). PBS replaced the primary antibodies in the negative controls. Secondary antibodies were conjugated to fluorescein isothiocyanate (FITC; 1:200; Chemicon AP192F, Temecula, CA, USA, and 1:100; SouthernBiotech, Birmingham, AL, USA). The specimens were counterstained with Hoechst (Sanofi, Shanghai, China). All the samples were examined under a fluorescence microscope (Olympus Optical).
Flow Cytometry (FCM) Analysis
Approximately 2.5 × 105 third passage PDLSCs were fixed with 4% paraformaldehyde for 15 min and then incubated with primary antibodies for cluster of differentiation 146 (CD146; 1:100; R&D Systems, Minneapolis, MN, USA), CD105 (1:120; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and STRO-1 (1:100; eBioscience, San Diego, CA, USA) at room temperature for 1 h, followed by fluorescein-conjugated secondary antibody at room temperature in the dark for 45 min. The percentage of PDLSCs staining positive for CD146 and CD105 was assessed using a FACS Calibur flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA).
Culture of PDLSC Sheet
The PDLSCs were seeded at an initial density of about 2.5 × 104/cm2 into culture plates and incubated for 48 h. Then the medium was changed to cell sheet culture medium (SCM), which consisted of DMEM supplemented with 10% fetal bovine serum (FBS), 0.292 mg/ ml glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml ascorbic acid. SCM was replaced every 3 to 4 days for a further 10 days (40). A mono-layer cell sheet (MCS) was lightly detached from the bottom of the culture plate with a cell scraper (NUNC, Rochester, NY, USA) and overlaid onto an undetached cell sheet and smoothed with forceps (Vitus, Qdong, China) to ensure the effective contact of two cell sheets. This was repeated a minimum of three times to generate a multilayered cell sheet. The multilayered cell sheet was cultured for 4 days.
Culture of Monolayer Cell Pellets (MCPs) and the Multilayer Cell Pellets (MUCPs)
After washing three times in the culture medium, the monolayer or multilayer cell sheets were transferred to a 10-ml conical polypropylene tube (Asahi Techno Glass Corp., Tokyo, Japan) and incubated with the SCM. The caps of the tubes were loosened to permit gas exchange, and the tubes were maintained at 37°C in a humidified atmosphere of 5% carbon dioxide. Within 12–24 h of incubation, the cell sheet began to shrink and formed a cell mass that did not adhere to the walls of the tube. Changes in the medium were carried out by observation at 24-h intervals, and the MCPs or MUCPs were harvested at time points up to 10 days.
Histological Analysis of MCPs or MUCPs
The MCS, bilayered cell sheet, MCPs, or MUCPs were fixed in 10% neutral buffered formalin (Keshi), dehydrated in a series of ethanol, embedded in paraffin, and cut into 5-mm sections. After deparaffinization, the sections were evaluated via routine hematoxylin and eosin (H&E; Baso, Zhuhai, China) staining according to the manufacturer's recommended protocol. All the samples were examined under a compound microscope (Olympus Optical).
Immunostaining of the MCPs or MUCPs
MCPs and MUCPs were fixed in 4% paraformaldehyde at 37°C for 15 min, washed three times, and then stained with primary antibodies for COL-I (1:100), COL-III (1:100; Abcam), fibronectin (1:100), and laminin (1:100). PBS replaced the primary antibodies in the negative controls. Secondary antibodies were conjugated to FITC (1:100) and rhodamine (1:200; Chemicon AP182F). The specimens were counterstained with Hoechst. All samples were examined under a fluorescence microscope.
Real-Time PCR
Total RNA was extracted from MCPs or MUCPs using TRIzol reagent (Invitrogen). First-strand cDNA synthesis was performed using PrimeScript® RT reagent kit (Takara, Shiga, Japan). Quantitect Sybr Green Kit (Toyobo, Osaka, Japan) and ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) were used for real-time PCR analysis. Primer sequences for COL-I, COL-III, fibronectin, laminin, and β-actin are listed in Table 1. The cycling conditions were as follows: 95°C for 10 min, 45 cycles at 95°C for 15 s, and 60°C for 1 min. The experiments were repeated three times, and mRNA levels were normalized to β-actin.
Oligonucleotide Primer Sequences Utilized in Real-Time PCR

Western Blot Analysis
Proteins were extracted from cell pellets and subjected to sodium dodecyl sulfate polyacrylamide gel electropho-resis (SDS-PAGE; Bio-Rad) and transferred to membranes (Bio-Rad) that were probed with primary antibodies for COL-I (1:100), COL-III (1:100), fibronectin (1:100), and laminin (1:100). Following several washes in Tris-buffered saline (Bio-Rad) with 0.2% Tween (Amersco, Solon, OH, USA), the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit or mouse secondary antibody (1:5,000; Zhongshanjinqiao, Beijing, China) and then developed using ECL agent (Millipore, Billerica, MA, USA).
Cell Adhesion and Migration Assays
The peripheral edges of the cell pellets were trimmed, and the resultant pellet was separated into halves using a stainless steel screen (40 mesh, from the cell dissociation sieve-tissue grinder kit, CD1-1KT, Sigma). The fragments were lightly detached from the bottom of the culture plate with cell scraper, resuspended in 500 ml SCM, and immediately seeded onto the 24-well plates or the TDMs, with the dissociated cells (DCs) and the MCS fragments as the control (n = 10). Changes in morphology of the MCS fragments with time were investigated by means of a live cell imaging system (Leica) and scanning electron microscope (SEM; FEI, Eindhoven, Netherlands) every day for up to 5 days (n = 10).
For the live cell imaging system observation, after the MUCPs or MCPs were seeded onto a dish (Corning), then SCM was injected gently along the side of the well until the MUCPs and MCPs were immersed. The plates were subsequently incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The cell pellet and plate interface was consecutively photographed at 5-min intervals. The whole process lasted for 5 h. Following this procedure, all images were observed, and the times required for the cells to migrate from the cell pellet were evaluated.
For SEM evaluation, the samples were washed in PBS three times, fixed with 2.5% glutaraldehyde (Keshi) at 0°C, dehydrated and dried in a critical point dryer (Tousimis, Chicago, IL, USA), and finally observed using SEM.
Cell Proliferation Assay
MCPs and MUCPs were seeded onto six-well plates and cultured in SCM for 3 days. The culture medium was then changed into SCM containing 30 μg/L bromode-oxyuridine (BrdU; Abcam), and the cells were cultured for 4 days. The cell pellets were harvested, washed three times with PBS, and then fixed in 4% paraformaldehyde for 30 min. The fixed cell pellets were denatured in 2 mol/L HCl for 60 min at 37°C and neutralized with 0.1 mol/L sodium borate (pH 8.5; Keshi) for 10 min. Immunohistochemical detection of BrdU was performed according to the manufacturer's protocol using a monoclonal mouse anti-BrdU antibody (1:180 dilution, Millipore, Billerica, MA, USA). Secondary antibodies were conjugated to FITC (1:200; Chemicon AP192F), and the specimens were counter-stained using Hoechst staining. All samples were examined using a fluorescence microscope (Olympus Optical). The cells that stained positive for BrdU per visual field (×10 ×20) were counted and analyzed.
In Vitro Coculture of the MCPs or MUCPs with the TDM
The preparation for TDM (size: 2 mm × 2 mm × 1 mm) and cell pellets were the same as described above. The cell pellets were reshaped (approximately 3 × 3 mm) at a seeding density of the fragment approximately 5 × 104 cells/cm2 and placed onto the surface of TDM in a six-well plate to construct the hybrids. The hybrids were incubated in a small volume of culture medium (0.5 ml α-MEM) at 37°C for 2 h to allow stable adhesion, and then, 1.5 ml α-MEM was added along the wall of the six-well plate. The media were changed every 2 days. The dissociated PDLSCs (at the same cell density) were used as a control. The hybrids were incubated in a six-well plate for 2 weeks, then washed in PBS three times, fixed with 2.5% glutaraldehyde at 0°C, dehydrated, dried in a critical point dryer, and finally observed under a SEM. The experiment was repeated at least three times.
In Vivo Transplantation of MCPs or MUCPs Into the Omental Pouch
All the operations were performed under general anesthesia. Explants of MCPs or MUCPs combined with or without TDMs were transferred to a culture dish and incubated at 37°C, 5% CO2, and 95% air atmosphere for 2 h and implanted into omental pouches for 4 weeks. Then all implants were harvested, fixed with 4% para-formaldehyde overnight at 4°C, decalcified with 10% EDTA (pH 8.0), and embedded in paraffin. Finally, paraffin sections (5 mm) were prepared for further histological study. Sections were stained with H&E according to the manufacturer's recommended protocols.
Generation of the Periodontal Defect Model and the In Vivo Transplantation of the MCPs and MUCPs
The rats were anesthetized with pentobarbital (Merok, Shanghai, China) to generate a periodontal defect model. After clinical assessment, a mucoperiosteal flap was raised, and alveolar bone was removed using a surgical burr (NSK, Tokyo, Japan) to create experimental periodontal defects in the mesial region of the maxillary first molars. The created alveolar bone defect was 1 mm in width, 4 mm in length, and 2 mm in depth (Fig. 1). The denuded root surface was rinsed with sterile water, treated with 17% EDTA for 3 min, and then rinsed again with water. MCS, MCPs, or MUCPs were transplanted into the periodontal defect.
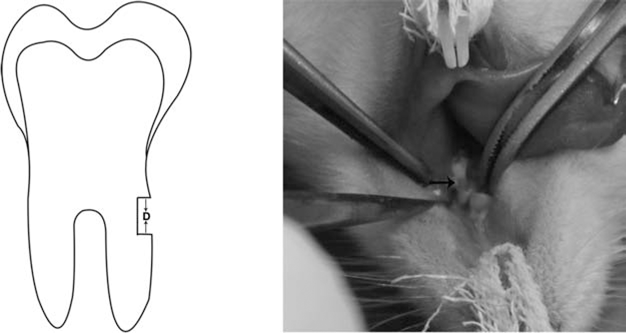
The diagrammatic illustration and image of the periodontal defect. The arrows (D) indicate the operative area.
Harvested samples were embedded in paraffin and sectioned (5 μm). Then these sections were respectively stained with H&E according to the manufacturer's recommended protocols. New mineralized matrix was analyzed quantitatively using an image analysis system (cellSens Dimension 1.4.1, Olympus).
Statistical Analysis
Data are expressed as means ± SD. Statistical significance was analyzed by SPSS 12.0 software (SPSS, San Rafael, CA, USA). Analysis of variance followed by Bonferroni-adjusted Student's t test was used to determine the significant differences between control and test group. Values of p < 0.05 were considered significant. All procedures were performed blindly.
Results
Isolation and Characterization of PDLSCs
The harvested PDLSCs were capable of forming colonies (Fig. 2A). Fifth passage PDLSCs still preserved the fibroblastic spindle shape (Fig. 2B). After 14 days of osteo-genic induction, PDLSCs were observed to form extensive mineralized nodules (Fig. 2C). They also showed the capability of adipogenic differentiation in adipogenic induction for 14 days (Fig. 2D). FACS analysis revealed that approximately 27.3% PDLSCs were positive for CD146, 11.1% for Stro-1, and 99.7% for CD105, which revealed rich mesenchymal stem cells existing in cultured cells (Fig. 2E). In addition, ICH showed that the PDLSCs were positive for COL-I, fibronectin, laminin, OCN, ALP, and ON (Fig. 2F).

Isolation and characterization of PDLSCs. (A) Periodontal ligament-derived stem cells (PDLSCs) formed a single colony when plated at a low density. (B) The fifth passage PDLSCs presented the satisfactory morphology. (C) When the third passage PDLSCs were cultured under osteogenic-inductive conditions for 4 weeks, mineralized nodules were found by Alizarin red staining. Scale bars: 1 mm. (D) Cultured third passage PDLSCs formed Oil red O-positive lipid clusters following 4 weeks of adipogenic induction. Scale bars: 1 mm. (E) Flow cytometric analysis of ex vivo expanded PDLSCs revealed the expression of negative control (1.5%), STRO-1 (11.1%), cluster of differentiation 146 (CD146; 27.3%), and CD105 (99.7%). (F) Immunocytochemical analysis showing that PDLSCs were positive for collagen I (COL-I), fibronectin, laminin, osteocalcin (OCN), alkaline phosphatase (ALP), and osteonectin (ON).
In Vitro Histological Examination of PDLSC Pellets
When PDLSCs were seeded into culture plates in DMEM plus 50 μg/ml ascorbic acid for 14 days, extensive extracellular matrix was laid down on the culture plate, and the MCPs were easily harvested from the edge of the culture dish to overlap another intact cell sheet (Fig. 3Ab). Furthermore, the MUCPs retained cell vitality in the center, and the sequential histological changes of the cell pellets showed the continuous increases in ECM, which was associated with no obvious fractures (Fig. 3Bf).

In vitro histological examination of PDLSC pellets. (A) Monolayer cell sheets (MCS) (a) cultured for 10 days formed a sphere and had rich extracellular matrix (ECM) with some fractures interspersed (b, c). Multilayered cell sheet (d) cultured for 10 days presented similarly abundant ECM, but no fractures were observed (e, f). Scale bar: 1 mm. (B) Histological assessment of the multilayer cell pellets (MUCPs) stained with hematoxylin and eosin. (a) MCS control exhibited less ECM. (b) The bilayered cell sheet exhibited a denser ECM after culturing for another day. (c) MUCPs showed a richer ECM after culturing for another 2 days. (d) Few fractures were observed in MUCPs cultured after 4 days. (e) The ECM became condensed, and the fractures were constricted in MUCPs cultivated using the 3D cultivation system for 8 days. (f) MUCPs condensed into an aggregate without fractures after 10 days of cultivation. Scale bar: 50 μm. (C) Immunocytochemistry (ICH) analysis showed that MCPs and MUCPs are positive for ECM molecules including COL-I, COL-III, fibronectin, and laminin. Scale bar: 100 μm. Real-time PCR (D) and Western blot analysis (E) of monolayer cell pellets (MCPs) and MUCPs showing that MUCPs contained higher levels of these ECM molecules than MCPs. *p < 0.01, **p < 0.001.
Meanwhile, we compared the morphology and histology of the two cell pellets fabricated by different fabrication modes. The MUCPs (Fig. 3Ae, f and Bf) contained denser and more regularly aligned ECM microarchitecture than the MCPs (Fig. 3Ab, c). More importantly, the fractures interspersed were observed in all MCP samples (Fig. 3Ac).
ICH showed that MUCPs and MCPs were positive for the ECM molecules including COL-I, COL-III, fibronectin, and laminin (Fig. 3C). Real-time PCR (Fig. 3D) and Western blot analysis further demonstrated that the levels of these ECM molecules were higher in the MUCPs than in the MCPs (Fig. 3E).
Cell Adhesion and Migration Assays
Cell adhesion and migration assays showed that the dissociated PDLSCs were significantly slower to adhere to the plate (within 4 h, 210 ± 20 min) than MCS (within 2 h, 70 ± 10 min) (Fig. 4). MUCPs were quicker to adhere and migrate onto the plate (within 2 h, 75 ± 5 min) than MCPs (within 2 h, 90 ± 5 min) (Fig. 5). The rate of cell crawling and proliferation of the MUCPs was lower compared with MCS, but after 72 h of culture the cells in the wells seeded with the two fragments both reached confluence (upper three rows in Fig. 4). DCs proliferated slowly on the TDM, but MCS adhered to the surface and covered the matrix with the intensive mineral deposits very swiftly. MUCPs settled and spread on the matrix quickly (lower two rows in Fig. 4).
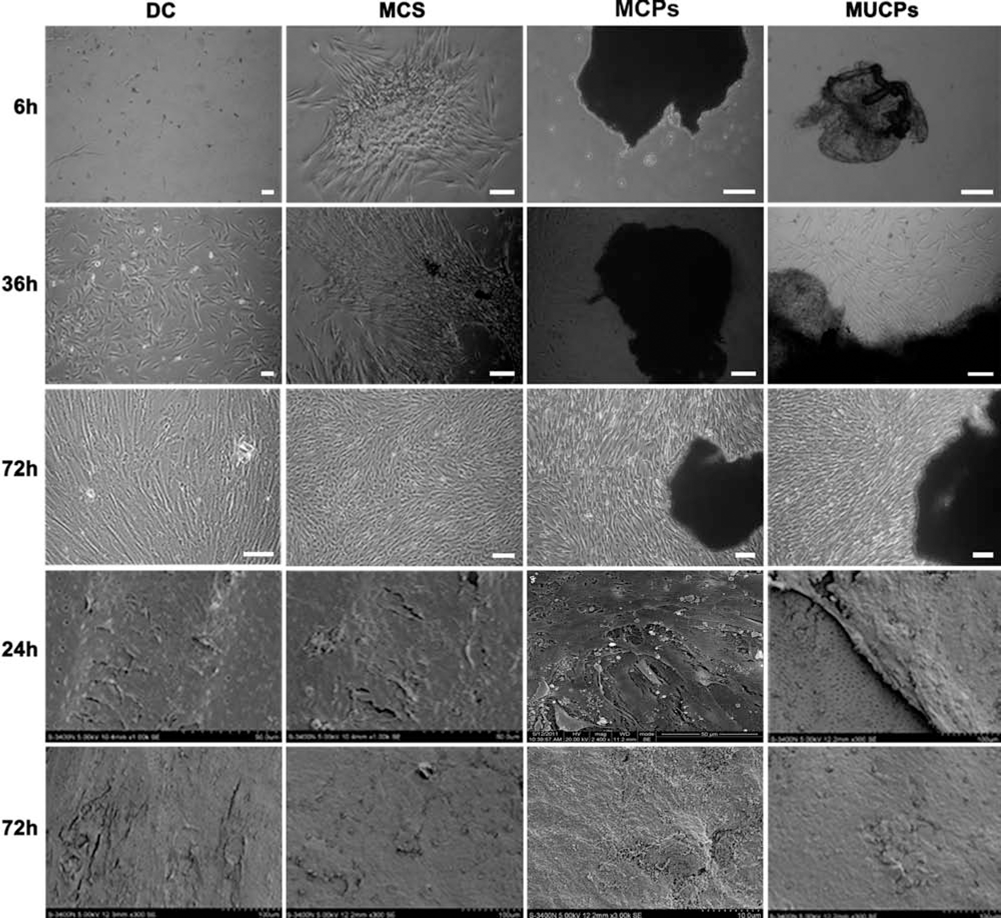
Increased cell attachment and proliferation in MUCPs compared with MCPs on plates (top three rows) and on TDM (bottom two rows). Dissociated cells (DC) showed lower settlement capability and proliferation rate. MCS adhered to the plate very rapidly and reached confluence within 3 days. MUCPs attached to plate swiftly, but cell scrawling was slower than DC and MCS and quicker than MCPs. Scale bar: 100 μm. DCs on the treated dentin matrix (TDM) showed a lower proliferation rate, but MCS adhered to the surface very swiftly and covered the matrix with the intensive mineral deposits. MUCPs and MCPs settled and spread on the matrix quickly.

The time required for MUCPs (A) and MCPs (B) to adhere and migrate onto cell culture plates. The cells began migrating from MUCPs at 75 min and MCPs at 90 min. White arrows show the adhesive and migrating cells.
In addition, we performed SEM analysis on the MUCPs and MCS seeded onto the TDM. After seeding the fragments onto the TDM for 72 h, we observed massive fiber attachment interspersed between the dentine surface and the cell sheet (Fig. 6B, C), formation of dense ECM and intimate cell junction, and mineral deposit after the delayed culture (Fig. 6D–F).
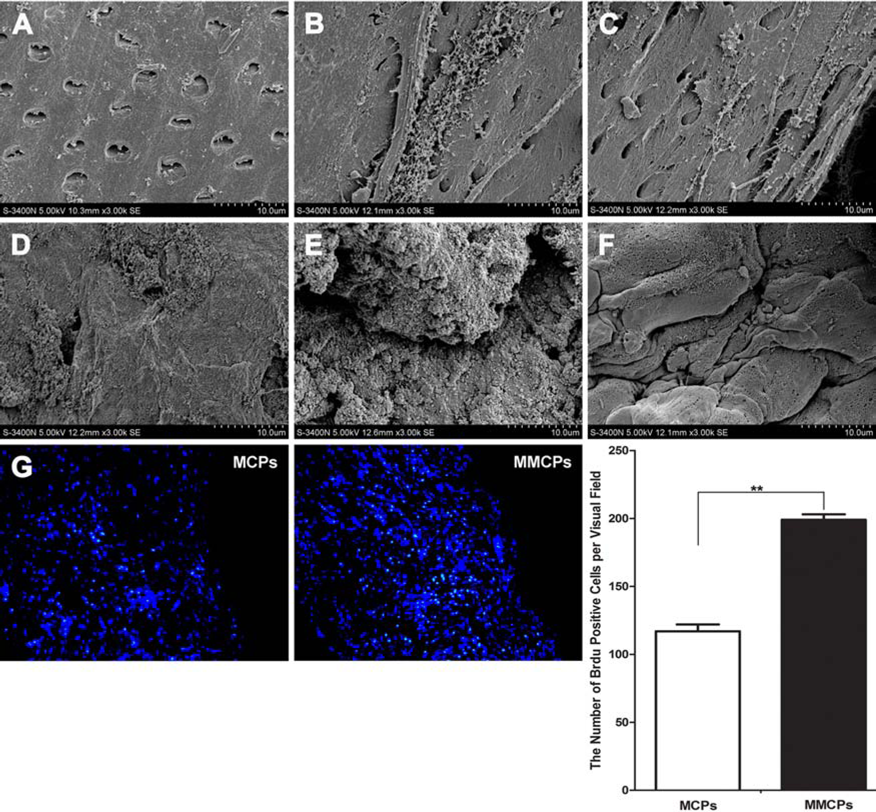
Scanning electron microscope (SEM) assessment of the structure of MUCPs and the interface of MUCPs with TDM. (A) TDM showed a clear surface and thoroughly exposed dentinal tubules. (B) The adhesion of the MUCPs on the TDM at day 1. (C) The linkage between the MUCPs and TDM on day 2. (D) Abundant mineral deposit in the MUCPs at day 5. (E) The high magnification of the intercellular linkage and ECM at day 5. (F) The cell–cell junction in MUCPs cultivated for 14 days. (G) Proliferation of cells in MUCPs and MCPs demonstrated by detecting bromodeoxyuridine (BrdU) expression. **p < 0.001.
Cell Proliferation Assay
BrdU incorporation assay showed that MUCPs had a higher proliferative capacity than MCPs (Fig. 6G).
Evaluating the Regenerative Capability of Cell Pellets Both In Vitro and In Vivo
To verify whether the PDLSC pellets maintained their differentiated state and had the potential to reconstruct the physiological architecture of cementum/periodontal ligament complex, we combined the cell sheet or cell pellet with the TDMs and cultured the hybrid for 14 days in vitro. The histological analysis indicated the aligned integration between the TDMs and the cell pellets. Notably, in the MUCP groups, obvious sandwich-like regenerated tissue was observed with newly deposited mineral and ECM linkage orderly filling the space between the cell pellets and surface of TDMs, while the regenerative changes in the MCP and MCS groups were comparatively indistinct (Fig. 7A).

The histological assessment of MUCPs, MCPs, and MCS in different host microenvironments. (A) The in vitro coculture of the hybrid of MUCPs, MCPs, or MCS with the TDM cultivated for 14 days. (B) Hematoxylin and eosin (H&E) analysis of grafts of the MUCPs, MCPs, and MCS without the support of TDM into the omental pouches of adult rats for 4 weeks. (C) The histological analysis of grafts of the MUCPs, MCPs, and MCS with the support of TDM into the omental pouches of adult rats for 4 weeks. (D) The histo-logical analysis of grafts of the MUCPs, MCPs, and MCS into the periodontal defects of adult rats for 4 weeks. M, mineralized matrix; E, extracellular matrix; N, notch of periodontal defect; C, cementum-like tissue; PDL, periodontal ligament. Scale bar: 100 μm.
Based on these in vitro findings, next we implanted the MCS and two kinds of cell pellets without TDMs into omental pouches of adult rats to evaluate their regenerative capability. After 4 weeks of incubation, all implants were harvested, and histology was analyzed. Implanted MCS, MUCPs, and MCPs all formed extensive fibers; however, the fiber alignment was more regular and bulky in the latter two groups. No obvious mineral deposit was observed in the three groups (Fig. 7B).
In contrast, the combination of the MCS and MUCPs/ MCPs with the TDMs implanted in the omental pouches of adult rats showed that the MUCPs/MCPs had the potential to completely reconstruct the physiological architecture of periodontal ligament/cementum complex. All 16 samples in MUCPs/MCPs and MCS were observed to form the heterogeneous amounts of periodontal fibers and cementum-like deposits (Fig. 7C).
Furthermore, we implanted the MUCPs/MCPs and the MCS into the periodontal defect of the rat model for 6 weeks. The longitudinal section analysis of the periodontal defect showed that the fibers inserted perpendicularly into the defect notch in the MUCPs/MCPs. The regeneration of the fiber and mineral deposit in the MCS was weaker than in the MUCPs/MCPs (Fig. 7C, D). Meanwhile, quantitative analysis indicated that MUCPs exhibited stronger mineralized tissue-forming capacity than MCPs during the periodontal defect tissue repair (Fig. 8).

Histomorphometric evaluation for regeneration of the mineralized tissue in periodontal defect model. The black dotted line indicates the boundary between regenerating mineralized tissue and host dentin. Quantitative evaluation showed more mineralized tissue formation in MUCPs than in MCPs. **p < 0.001.
Discussion
Cell sheet engineering for periodontal therapy requires suitable mechanical properties as well as vigorous cellular viability (8,15,21,25). The increase of the mechanical integrity of the sheet is generally dependent on stimulating the secretion of the endogenous ECM of the seed cells (17,18). We fabricated MUCPs with the aim to promote the secretion of endogenous ECM while maintaining cellular viability. We confirmed that cultured MUCPs contained higher levels of ECM molecules, such as COL-I, COL-III, fibronec-tin, and laminin, which implied that MUCPs have stronger mechanical properties than MCS. In this study, the multi-layered cell sheet was used to fabricate the MUCPs, instead of the previously reported MCS mode. By increasing the ECM deposition and the number of cell layers, the strength of an ECM scaffold can be increased. This is consistent with previous studies, which showed that by increasing the number of layers of small intestinal submucosa–extracellu-lar matrix (SIS-ECM) in a scaffold from two to four there was nearly 150% increase in strength (5,11). The 3D cultivated cell sheets contracted into tightly condensed spherical pellets mostly due to the contraction of the cytoskeletal elements and rich collagen fiber bundles. The collagen fibers in the cell pellets increased logistically and oriented more regularly, thus making it less resistant to stretch.
The ECM scaffold is believed to act as a supportive medium and delivery channel for the diffusion of nutrients from the blood to the cells embedded in the scaffold skeleton (14,30), but its potential is limited as it is confined by the dense collagen fiber architecture. Hence, the maximum number of cell layers in a sheet amid with viable cells was estimated to be less than five in a prior study (31). In the present study, we analyzed the histological changes in the MUCPs but did not observe obvious fractures within PDLSC pellets as previously described (40). This may be partly attributed to better integration between cell sheets given the rich ECM molecules contained in the MCPs. Abundant signal molecules residing on the surface of the MCPs facilitated the deposition and organization of new ECM and the interlinkage between the opposite cell sheets, which may enhance the biomechanical properties of the cell pellets and guarantee the nutrient supply to the cells in the center of the cell pellets.
ECM molecules contribute to the maintenance of the optical cellular function of the cell sheet (12,39). It is known that fibronectin modulates and maintains the composition and stability of the ECM and increases adhesion-dependent and -independent cell growth and cell contractility (23,37). Adhesion and proliferation are two key indicators of cell fate and behavior (19,38). In this study, we evaluated cell pellet adhesion and proliferation. Interestingly, the cells from MUCPs spread and proliferated more rapidly than those from MCPs. In the initial stage, the cells from the cell pellets spread on and attached to the plate and dentine surface slightly slower than the cells from MCS possibly because of the constraint of the dense collagen fibers. Functionally, cell pellets were combined with TDM and incubated for 2 weeks in vitro, in which obvious mineralization on the interface and interwoven fibers between the cell sheet and matrix were observed. Taken together, these results demonstrated that MUCPs facilitated the secretion of ECM and did not reduce cell vitality.
The mechanisms by which natural ECM scaffolds facilitate the constructive remodeling of tissues are still not completely understood (3,4). However, it has been verified that the ECM conveys intricate signals from the microen-vironment to modulate cell proliferation and differentiation during tissue development and regeneration. A precise understanding of the function of cell pellets can be obtained by transplanting cell pellets to the diverse recipient microen-vironment. The periodontal regeneration includes the soft tissue of the periodontium and hard tissue of cementum and bone (10,32). In this study, the PDLSC sheets and cell pellets were transplanted into the omentum sac and peri-odontal defect model. Without the induction and support of the TDM, the cell pellets formed extensive fibers without specific orientation. The combination of cell pellets with the TDM implanted in the omental pouches of adult rats showed complete periodontal ligament/cementum complex formation, which resembled the physiological architecture of periodontium. This evidence suggests that the 3D cultivation system may enhance the mineral capability of the PDLSCs and promote cementum deposition.
Notably, we found that the regenerated periodontal tissues by PDLSC pellet transplantation were similar in different recipient microenviroments. In this study, when the samples were examined histologically along the anatomical structure (vertically sectioned), regeneration of PDL was observed in all samples (10/10). However, when sections were examined horizontally from the same groups, the results were not as clear, and not all sections revealed the regeneration of periodontal tissue. For example, in 3 of 15 in situ grafts and 4 of 11 heterotopic grafts, the formation of PDL-like fibrous tissues and the deposits of new cementum-like tissue were incompletely regenerated. Favorable regenerative results depend on a close contact of the cell pellets with the matrix (2,21). The contraction of the ECM in the cell sheet mainly alters its shape and evenness and, meanwhile, changes surface tensile force and cell–cell interaction with cells in the pellets (19,28). MCS cannot be spread onto uneven matrix very easily (21,40), while tensile cell pellets can be inserted into the recipient site handily with the preferable biomechanics. Successful periodontal regeneration requires faster anchorage and proliferation of mesenchymal cells than that of the epithelial tissues to prevent the formation of a long junctional epithelium (9,33). The better adhesion and proliferation capability of the cell pellets enables the cells to preponderantly settle down onto the treated root surface. This may explain the high success rate from the cell pellets and reminds us that the successful postsurgical assessment of periodontal therapy by monodirectional histological observation is likely to give a false impression.
Although this study demonstrated that cell pellets significantly promoted periodontal regeneration, questions remain unanswered. For instance, it is unclear how PDLSCs in cell pellets differentiate and cross talk with cells from the host. Furthermore, it is not known how the matrix in the cell pellets regulates PDLSC differentiation. Further studies are required before the clinical potential of this periodontal regeneration strategy can be utilized to the fullest potential.
In summary, in this study we developed a novel fabrication method for constructing cell pellets. The MUCPs exhibited a rich ECM, better mechanical properties, and stronger cell viability. The 3D cell pellet was easily manipulated and allowed for the complete reconstruction of the physiological architecture of the cementum/ periodontal ligament complex. Future optimization and mechanistic studies of the cell pellets will be necessary to promote its development as a promising alternative to the periodontal defect repair for future clinical application.
Footnotes
Acknowledgments
This study was supported by grants from the Nature Science Foundation of China (30725042, 81020108019, and 31030033) and the National Basic Research Program (973 Program) (2010CB944800). The authors declare no conflicts of interest.