
Abstract
Harvesting, expansion, and directed differentiation of human bone marrow-derived mesenchymal stem cells (BM-MSCs) could provide an autologous source of surrogate β-cells that would alleviate the limitations of availability and/or allogenic rejection following pancreatic or islet transplantation. Bone marrow cells were obtained from three adult type 2 diabetic volunteers and three nondiabetic donors. After 3 days in culture, adherent MSCs were expanded for two passages. At passage 3, differentiation was carried out in a three-staged procedure. Cells were cultured in a glucose-rich medium containing several activation and growth factors. Cells were evaluated in vitro by flow cytometry, immunolabeling, RT-PCR, and human insulin and c-peptide release in responses to increasing glucose concentrations. One thousand cell clusters were inserted under the renal capsule of diabetic nude mice followed by monitoring of their diabetic status. At the end of differentiation, ~5–10% of cells were immunofluorescent for insulin, c-peptide or glucagon; insulin, and c-peptide were coexpressed. Nanogold immunolabeling for electron microscopy demonstrated the presence of c-peptide in the rough endoplasmic reticulum. Insulin-producing cells (IPCs) expressed transcription factors and genes of pancreatic hormones similar to those expressed by pancreatic islets. There was a stepwise increase in human insulin and c-peptide release by IPCs in response to increasing glucose concentrations. Transplantation of IPCs into nude diabetic mice resulted in control of their diabetic status for 3 months. The sera of IPC-transplanted mice contained human insulin and c-peptide but negligible levels of mouse insulin. When the IPC-bearing kidneys were removed, rapid return of diabetic state was noted. BM-MSCs from diabetic and nondiabetic human subjects could be differentiated without genetic manipulation to form IPCs that, when transplanted, could maintain euglycemia in diabetic mice for 3 months. Optimization of the culture conditions are required to improve the yield of IPCs and their functional performance.
Keywords
Introduction
Diabetes mellitus is a wide-spread devastating disease affecting millions of people worldwide. Maintaining a good glycemic control with exogenous insulin imposes a burden on patients. Transplantation of intact pancreas or isolated pancreatic islets are alternative treatments for patients with type 1 diabetes. However, the shortage of cadaveric organs and the necessity to use immunosuppression are limiting factors (35).
Recent progress in the field of regenerative therapies has focused attention on generation of surrogate β-cells from stem cells derived from embryonic, umbilical cord blood, Wharton's Jelly, and a variety of adult tissues (4). Human embryonic stem cells (h-ESCs) can be expanded in the undifferentiated state and differentiated to all cell types including insulin-producing cells (IPCs) (22, 24, 36). So far, the use of these cells is burdened by ethical considerations as well as by practical problems: the lack of available embryos, difficulties with the generation of immunocompatible cells, and the risk of uncontrolled malignant proliferation of residual undifferentiated cells. Insulin-producing cells can also be obtained by directed molecular engineering of umbilical cord stromal mesenchymal stem cells (MSCs) (43). These sources have the advantage of a large potential donor pool. Nevertheless, the risks of rejection and the need for immunosuppression remain.
Adult stem cells have been identified in many organs and tissues including the bone marrow. Bone marrow (BM) MSCs are pluripotent and can differentiate into lineages of mesenchymal, endodermal, and ectodermal cells including osteoplasts, adipocytes, myoblasts, and endocrine cells (31). This source provides the opportunity for generation of large numbers of β-cells without molecular engineering. Harvesting, expansion, and directed differentiation of autologous stem cells would alleviate the major limitations of availability and/or allogenic rejection. Several investigators tried to induce bone marrow-derived MSCs of rodents to generate IPCs (7, 16, 44). In a previous study, we had successfully treated diabetic Sprague–Dawley rats with islet-like clusters derived from bone marrow of inbred donors (13). In an in vitro study, Sun et al. partially characterized differentiated MSCs from human diabetic subjects (42). Herein, we report our studies to generate well-characterized endocrine cells from adult human bone marrow-derived MSCs from nondiabetic and type 2 diabetic subjects and test their insulin secretory potential in a mouse model of diabetes.
Materials and Methods
Retrieval of Human Bone Marrow Cells
The required approvals for this study were obtained from the ethical committee of the University of Mansoura. Bone marrow aspirates (BMA) were collected in heparin from six consenting donors. Three donors were type 2 diabetic patients requiring insulin from whom samples were obtained from the iliac crest, and three were nondiabetic volunteers from whom samples were obtained during hip replacement surgery (Table 1).
Characteristics of the Bone Marrow Volunteers

DM, diabetes mellitus; Hba1c, glycosylated hemoglobin.
Isolation, Expansion, and Differentiation of Bone Marrow-Derived MSCs
The BMA were diluted 1:1 with low glucose Dulbecco's modified Eagle's medium (DMEM, Sigma, St. Louis, MO, USA), layered on top of a density gradient (Ficoll-Paque, 1.077 g/ml) (Pharmacia, Uppsala, Sweden), and centrifuged for 20 min at 600 × g. Cells were collected from the media/ Ficoll interface, washed twice in phosphate-buffered saline (PBS), and resuspended in 10 ml of low glucose complete DMEM supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 U/ml penicillin, and 100 U/ml streptomycin (Sigma). One milliliter of the BMA yielded ~1.5 × 106 nucleated cells. The collected cells were cultured in complete DMEM at a density of 5 × 105 cells / ml (10 ml in 25 cm2 tissue culture flask) and incubated at 37°C in 5% CO2 incubator. Aliquots were frozen in liquid nitrogen for subsequent expansion and examination.
After 3 days, the nonadherent cells were discarded. The remaining adherent MSCs were cultured to 80% confluence before passaging with trypsin. Cells were resuspended with complete DMEM and replated at a ratio of 1:2 and cultured for a further 8 days. This step was repeated for a second passage. The cells then had the appearance of fibroblast-like cells.
Differentiation of the MSCs to form endocrine cells: At passage 3, MSCs were cultured at a density of 1 × 105 cells/ml in serum-free, glucose-rich DMEM (25 mmol/L) containing 0.5 mmol/L β-mercaptoethanol (Sigma) and incubated for 2 days. The medium was replaced with serum-free, glucose-rich medium containing 1% nonessential amino acids (Sigma), 20 ng/ml basic fibroblast growth factor (Sigma), 20 ng/ml epidermal growth factor (Sigma), 2% B27 supplement (Gibco BRL, Life Technologies, UK), and 2 mmol/L l-glutamine (Sigma) and cultured for a further 8 days. Finally, the cells were cultured for an additional 8 days in serum-free, glucose-rich DMEM containing 10 ng/ ml β-cellulin (Sigma), 10 ng/ml activin A (Sigma), 2% B27 supplement, and 10 mmol/L nicotinamide (Sigma).
Evaluation of the Cells
Characterization of Isolated MSCs by Flow Cytometry
For flow cytometric analysis, the bone marrow-derived MSCs at passage 3 were trypsinized, centrifuged at 300xg for 8 min, and resuspended in PBS at a concentration of 1 × 106 cells/ml. One hundred microliters of aliquots were labeled (30 min) with antibodies against CD14, CD45 fluorescein isothiocyanate (FITC) or CD73, CD34 phycoerythrin (PE) (Becton-Dickinson, USA), or CD105 PE or CD90 FITC (Becton-Dickinson, USA); washed with 1 ml of stain buffer (BD-Pharmingen, USA); and resuspended in 500 μl of stain buffer. The labeled cells were analyzed using an argon ion laser with a wave length of 488 nm (FACS Calibur, Becton-Dickinson, USA). A total of 10,000 events were obtained and analyzed with the Cell Quest software program (Becton-Dickinson, USA). Control staining with appropriate isotype-matched monoclonal antibodies was included.
Gene Expression by RT-PCR
Total RNA was extracted from undifferentiated MSCs at passage 3 and differentiating cells at days 2, 10, and 22 into TRIzol (Invitrogen Corporation, Grand Island, NY, USA). Lung cells (WI 38 cell line; ATCC CCL 75; Manassas, VA, USA) and human pancreatic islets were included as a negative and positive control, respectively. Expression of a variety of genes was examined (Table 2). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was also included as an internal control. Briefly, 1 μg of total RNA was converted to cDNA using high-capacity cDNA archive kit (ABI PRISM 3100 Genetic Analyzer, Applied Biosystems, Foster City, CA, USA). Then 2 μl of the cDNA was amplified using 25 pmol of each primer pair, 12.5 μl of 2x Taq PCR Master Mix (QIAGEN Inc., Valencia, CA, USA), and nuclease-free water to a total volume of 25 μl. The cycling parameters of the PCR amplification were as follows: initial denaturation at 95°C for 5 min, followed by 30 cycles of amplification and final extension at 72°C for 10 min. The PCR products were electrophoresed in 1% agarose gel (Sigma) and visualized by ethidium bromide staining (Sigma). In addition, relative quantitative RT-PCR (qPCR) was carried out for undifferentiated MSCs and at full differentiation (22 days). Human islets served as a positive control. The test was carried out using Stell ARray Gene Expression System (Lonza, Walkersville, MD, USA), which includes CD44. Amplifications were performed in a 20-μl reaction volume containing 10 μl 2x SYBR Green Master Mix (Takara BioINS, CA, USA). Reactions were performed on a 7000 Real-Time PCR System (ABI PRISM, Applied Biosystem, CA, USA). A model introduced by Pfaffl was used for calculation (30).
List of Human Gene-Specific Primers in RT-PCR

Pdxl, pancreatic and duodenal homeobox 1; MafA, v-maf musculoaponeurotic fibrosarcoma oncogene homolog A; MafB, v-maf musculoaponeurotic fibrosarcoma oncogene homolog B; Pax4, paired box 4; Ngn3, neurogenin 3; NeuroD1, neurogenic differentiation 1; Rfx6, regulatory factor X 6; GLUT-2, glucose transporter member 2; PC1, proprotein convertase subtilisin/kexin type 1; PC2, proprotein convertase subtilisin/kexin type 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; bp, base pair.
Immunolabeling
Cell preparations at different stages of the differentiation protocol and engrafted IPCs in the kidneys of mice were immunolabeled for insulin (rabbit monoclonal, Cell Signaling Technology, Danvers, MA, USA), glucagon (rabbit polyclonal anti-glucagon, Cell Signaling Technology), rabbit anti-human somatostatin (DakoCytomation, Glostrup, Denmark), or human c-peptide (rabbit polyclonal; Cell Signaling Technology). Cell culture specimens in situ on chamber slides (Nunc, Rochester, NY, USA) were fixed in 4% paraformaldehyde, and tissue was fixed in 2.4% formaldehyde and prepared in wax. Secondary antibodies employed were swine anti-rabbit immunoglobulin labeled with FITC, antimouse TRITC (tetramethyl rhodamine isothiocyanate; DakoCytomation), or biotin avidin complex and DAB. Cell viability was examined using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) detection kit (GenScript, USA) and the frequency of cell division by labeling for Ki67 (Cell Marque, USA). Preliminary confocal images to determine colocalization of hormones were captured after labeling for insulin (inhouse guinea-pig antibody to human insulin B-chain) and glucagon (mouse monoclonal antibody; Sigma).
For nanogold immunostaining, the IPCs were fixed in 2% formalin and 0.5% glutaraldehyde (both EM grade from Ted Pella) in phosphate-buffered saline (PBS) overnight at 4°C. After washing 3 (15 min with phosphate-buffered saline (PBS) containing 0.05% Tween 20 (Bio-Rad Laboratories, Hercules, CA, USA), they were blocked for 6 h in casein blocking buffer (BioRad) with 0.05% Tween 20 at room temperature. Subsequently, samples were incubated with mouse monoclonal anti-c-peptide (1H8) antibody (Abcam, ab8297) at 1:50 dilution in blocking buffer, overnight at 4°C. On the next day, samples were washed for several hours with PBS–0.05% Tween 20 and incubated overnight at 4°C with anti-mouse antibody conjugated with 1.4 nm gold (Nanoprobes, Yaphank, NY, USA) diluted at 1:50 with blocking buffer. On the next day, samples were washed 3 (15 min with PBS–0.05% Tween 20, postfixed for 20 min in 1% glutaraldehyde in PBS–0.05% Tween 20, washed 3 (15 min in water, silver enhanced, and either photographed as a whole mount or processed for electron microscopy without the osmium tetraoxide treatment, as described previously. Whole-mount immunostained IPCs were contrasted with 0.5% uranyl acetate only (without osmium tetraoxide) and embedded in Epon. Thin (70–100 nm) sections were examined and photographed in JEOL 1200 transmission electron microscope (1, 19, 20). Undifferentiated MSCs as well as human islet cells were similarly labeled to serve as negative and positive controls, respectively.
Determination of In Vitro Insulin and c-Peptide Release in Response to Increasing Glucose Concentrations and Peptide Content of Cell Preparations
Four different samples (1 million cells) were collected from the same batch of each donor at the end of the differentiation period for measurement of hormones released. Cells were initially incubated for 3 h in glucose-free Krebs–Ringer bicarbonate buffer (KRB). This was followed by incubation for 1 h in 3.0 ml of KRB containing 5.5, 12, or 25 mM glucose concentrations. The supernatant was collected at the end of each incubation period. The collected samples were frozen at −70°C until assayed using an Elisa kit with a minimum detection limit of 1.76 μIU/ml (DRG Diagnostic, Germany).
Insulin and c-peptide content were determined in cells that had been cultured in 25 mM glucose. These were washed three times with PBS and then suspended in 50 mM of HCL/70% ethanol. Following centrifugation at 12,000 × g for 5 min, the supernatant was collected, neutralized by adding 50 mM NaOH, and stored at −70°C for ELISA assay. Once the insulin and c-peptide content were determined, the cells were homogenized in chilled PBS. The homogenate was then centrifuged at 1,700 × g for 10 min at 4°C and then at 28,000 × g for 20 min at the same temperature. The supernatant was analyzed for its protein content using the Bradford method.
In Vivo Transplantation Studies in Mice
The ability of the differentiated cells to establish normoglycemia in diabetic nude mice (Swiss Nu/Nu, Charles River Laboratories, Paris, France) was examined by their implantation in the renal subcapsular space. Approval for these experiments was obtained from the ethical committee of the University of Mansoura. Diabetes was chemically induced by a single dose of 220 mg/kg of streptozotocin (STZ, Sigma). The mice were considered diabetic once the blood glucose levels exceeded 16.5 mmol/L for two consecutive readings. Fifty animals were utilized (18 males and 32 females with an average age of 12 weeks). These were divided into four groups: normal mice (n = 10: 3 males and 7 females), diabetic mice treated with culture media only (n = 10: 4 males and 6 females), diabetic mice treated with 1 million undifferentiated MSCs (n = 10: 4 males and 6 females), and diabetic mice treated with differentiated cells generated from MSCs of donor (1) (n = 20: 8 males and 12 females). In anesthetized mice, 1,000 IPC clusters, equivalent to 1 million cells, in 20 μl of culture media were implanted in each mouse.
The biochemical profiles of the mice in all groups were periodically assessed. Blood samples were obtained from the tail vein and measured for blood glucose, serum mouse insulin, human insulin, and c-peptide by ELISA (DRG Diagnostic, Germany). At the end of the observation period, an oral glucose tolerance test was carried out. Glucose (1 g/kg) was administered orally by gavage to the overnight fasted mice. Blood samples from the tail vein were measured before and 15, 30, 60, 90, and 120 min after glucose administration using glucometer strips (Accu-Check, Roche Diagnostics, Bazle, Switzerland).
Three months after transplantation, nephrectomy was carried out for the cell-bearing kidneys. Kidneys were prepared for histological studies to examine the grafts. The blood glucose and human insulin levels of the surviving animals were monitored.
Statistical Analysis
Nonparametric data were evaluated by Friedman's test. Post hoc testing was performed by the Wilcoxon's Signed Ranking, and the p values were corrected by Bonferroni adjustments. A value of p < 0.05 was considered significant.
Results
Morphological and Phenotypical Characterization of the Cultured MSCs
At the end of the expansion phase, the cells became homogenous, spindle shaped, fibroblast-like, and arranged in monolayers. Flow cytometric analysis showed that they expressed high levels of CD73, CD90, and CD105, but negligible levels of CD14, CD34, and CD45 (Table 3). These results indicate that majority of the bone marrow-derived cells were MSCs. From a starting culture of 3 × 106 MSCs, ~5,000 clusters (approximately 5 million cells) were present at the end of the differentiation procedure. During the differentiation phase, the cells gathered gradually in groups with the formation of three-dimensional aggregates. At the end of the differentiation protocol, the cells formed clusters with a spheroidal morphology (Fig. 1). No apparent differences in the rate of growth or differentiation was observed between different cell batches from the same donor or between cells from different donors with or without diabetes.

Morphological changes of mesenchymal stem cells (MSCs) during differentiation. (A) Undifferentiated MSCs, at the end of the expansion phase. (B) Formation of cell aggregates at day 10. (C) Forrmation of cell clusters at day 18. (D) Collected clusters.
Flowcytometric Quantitation of Surface Markers of the Undifferentiated MSCs

At the end of differentiation, immunofluorescent labeling demonstrated that approximately 5–9% of cells were hormone positive both in the monolayers and in the cell aggregates. Insulin, c-peptide, and glucagon appeared as punctuate labeling within the cytoplasm. Furthermore, insulin and c-peptide were coexpressed (Fig. 2). The c-peptide-positive cells were TUNEL negative. In contrast to the undifferentiated MSCs, differentiated cells were Ki67 negative (Fig. 3). By electron microscopy, nanogold immunolabeling showed the presence of c-peptide reactivity at the endoplasmic reticulum of the IPCs and the islet cells (Fig. 4A, B). The cytoplasm of the undifferentiated cells was unlabeled (Fig. 4C).

Immunofluorescent staining of differentiated insulin-producing cells (IPCs). (a) Positive staining for insulin (green) with counter staining for DAPI (arrow). (b) Positive staining for c-peptide (red) with counter staining for DAPI (arrow). (c) Electronic merge of insulin and c-peptide. Coexpression of insulin and c-peptide (yellow) by the same cells could be seen (arrow).

Immunohistochemical staining for the proliferation marker Ki67. (A) The undifferentiated MSCs were positive for staining with Ki67. (B) The differentiated IPCs were negative for staining with Ki67.

Immunostaining with nanogold. (A) Ultrastructure analysis of the differentiated IPCs shows concentration of c-peptide (arrows) at the cisternae of rough endoplasmic reticulum. (B) Fragment of pancreatic islet cells shows c-peptide labeling in the vicinity of rough endoplasmic reticulum (positive control). (C) Fragment of undifferentiated cell shows unlabeled cytoplasm for c-peptide (negative control). RER, rough endoplasmic reticulum; m, mitochondria.
Gene Expression During and After Differentiation
Gene expression profiles throughout the differentiation process were examined in all donors. Expression of genes for pancreatic endocrine development was not detected in the undifferentiated MSCs. Two days after induction of differentiation, markers for the transcription factors (Pdx1, MafB, Pax4) were expressed (see Table 2 for gene definitions). After 10 days of differentiation, in addition to the aforementioned genes, markers for the transcription factors MafA, Ngn3, NeuroD1; the glucose transporter GLUT-2; and pancreatic islet enzymes PC1 and PC2 were expressed. Shortly thereafter, expression of Rfx6 was noted. Finally, there was full expression of the endocrine genes: insulin, glucagon, and somatostatin. Expression for pancreatic enzyme, glucokinase (GCK) was also noted, while gene expressions for nestin and Ngn3 were extinguished (Fig. 5A). A preliminary observation of qPCR for some genes in cells from donor [1] demonstrated that, at 22 days after differentiation, Pdx1 expression was between 1% and 10% of that in human islets. Insulin gene expression was lower at <1% of that in human islets (Fig. 5B).

Gene expression profile. (A) RT-PCR of the undifferentiated MSCs and during the various phases of their differentiation (days 2, 10, and 22). At the end of differentiation (day 22), the gene expression of IPCs was similar to that of human islets. Both expressed transcription factors (Pdx1, MafA, MafB, Pax4, Rfx6, and NeuroD1), pancreatic enzymes (GK, PC1, and PC2), and endocrine hormones (insulin, glucagon, and somatostatin) (see Table 2 for gene definitions). (B) qPCR: undifferentiated cells did not express any transcription factors or genes of endocrine hormones. At full differentiation (22 days), Pdx1 expression was between 1% and 10% of that of human islets. Insulin gene expression was <1% of that of human islets.
In Vitro Human Insulin and c-Peptide Release in Response to a Glucose Challenge
The differentiated cells released increasing amounts of insulin and c-peptide in response to increasing glucose concentrations (p < 0.05). The insulin but not the c-peptide content in cells derived from diabetic donors was significantly higher than that of cells from nondiabetic donors (p < 0.05) (Table 4).
In Vitro Human Insulin Release and Content
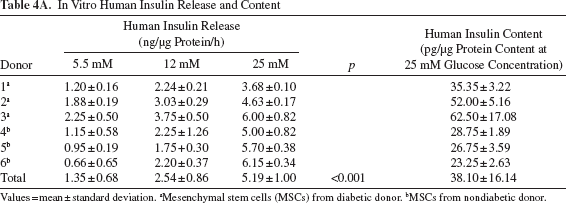
Values = mean ± standard deviation.
Mesenchymal stem cells (MSCs) from diabetic donor.
MSCs from nondiabetic donor.
In Vitro Human c-Peptide Release and Content
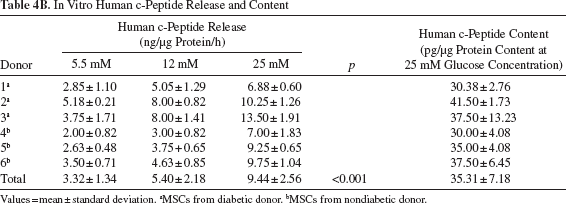
Values = mean ± standard deviation.
MSCs from diabetic donor.
MSCs from nondiabetic donor.
Outcomes of In Vivo Transplantation Experiments
The blood glucose levels of the diabetic mice implanted with the differentiated clusters of cells were normalized within few days, whereas those receiving no cells or undifferentiated cells remained hyperglycemic. Three mice that received differentiated cells died 2, 3, and 8 weeks posttransplantation. The remaining 17 animals remained euglycemic throughout the observation period of 3 months (Fig. 6A). Serum levels of human insulin and human c-peptide were measurable and maintained during the same period (Fig. 6B, C). Serum levels of mouse insulin were negligible in comparison to normal mice (Fig. 6D). At the end of the observation period, glucose tolerance curves in the IPC-transplanted mice were not significantly different to that of normal controls (Fig. 7). Eight animals died during nephrectomy of the cluster-bearing kidneys. The surviving nine mice became hyperglycemic, and the human insulin levels in their sera dropped to zero. These animals died a few days after the procedure. The islets of their native pancreata were negative for insulin by immunohistochemistry. No difference in results was observed relative to the sex of the recipient mice. Histology of the removed kidneys revealed the presence of cell clusters under the renal capsule. These clusters contained some cells coexpressing insulin and c-peptide or were glucagon positive. These immunoreactive cells were largely situated at the periphery of the clusters. Confocal microscopy with dual labeling for insulin and glucagon demonstrated that these hormones were in different cell types (Fig. 8).

Biochemical profile of diabetic nude mice treated by implantation of IPCs under the renal capsule (green). The blood glucose levels (A) were normalized few days after implantation and maintained throughout the observation period. The diabetic status resumed following nephrectomy of the IPC-bearing kidneys. Serum human insulin (B) and c-peptide (C) became measurable after implantation and rapidly disappeared following nephrectomy of the IPC-bearing kidneys. On the other hand, serum mouse insulin (D) was negligible.

The oral glucose tolerance tests. Transplanted animals (green), normal animals (red), and diabetic animals (blue). The curve of the IPC-transplanted animals approximated that of normal controls.

Histology of the removed IPC-bearing kidneys. Hematoxylin and eosin examination (A) demonstrates the implanted IPCs under the renal capsule (arrows). Positive immunofluorescent staining for insulin (arrow) (b) and c-peptide (arrow) (c). Electronic merging reveals coexpression of insulin and c-peptide by the same cells (arrow) (d). Confocal study with dual labeling for insulin (red) and glucagon (green) was positive in the IPCs under the renal capsule (e) and in human islets (f). These two hormones were localized in different cell populations (arrows).
Discussion
The ability to purify, culture, and manipulate multipotent stem cells from nonembryonic origin could provide a source to replace tissues for the treatment of degenerative disorders. Several in vitro studies have shown that bone marrow-derived stem cells of murine origin could be reprogrammed under certain culture conditions to become functional insulin-producing cells (6, 7, 16, 29). Other studies had provided data indicating that human bone marrow-derived MSCs can express insulin and key transcription factors of the endocrine pancreas developmental pathway upon genetic and/or microenvironmental manipulation in vitro (18, 27).
The current study reports a potential source to generate IPCs from human BM-MSCs by directed differentiation using soluble factors in culture media. We have opted to use MSCs for several reasons. MSCs residing in the bone marrow are multipotent and can differentiate into lineages of mesenchymal, endodermal, and ectodermal cells (18, 31). Evidence had also been provided that MSCs could be expanded ex vivo for more than 50 population doublings without obvious signs of differentiation (33). Accordingly, they can provide a rich source of autologous stem cells. An additional feature of bone marrow MSCs is their lack of expression of class II antigens and their ability to exert immunosuppressive activity on T cells (9). These properties suggest their possible use for the treatment of type 1 diabetes mellitus by abrogation of the associated autoimmunity and might also allow the use of allogenic MSCs if autologous grafts could not be used (8, 12).
We have shown that MSCs derived from human adult bone marrow of both diabetic and nondiabetic donors can be purified on the basis of their ability to adhere to plastic. The cells that had assumed spindle-shaped morphology at the end of expansion were negative for CD14, CD34, and CD45, which indicates that they were not hematopoietic stem cells and positive for CD73, CD90, and CD105, markers for MSCs (10).
Different protocols have been tried to induce bone marrow-derived MSCs to differentiate into IPCs in vitro (7, 16, 42). High glucose concentration was considered as a potent inducer for pancreatic islet differentiation (2). Initially, β-mercaptoethanol was added to the culture medium to induce expression of Pdx1 (44). Subsequently, fibroblast and the epidermal growth factors were used, since these were shown in an experimental model to stimulate proinsulin biosynthesis and 3H-thymidine incorporation (5). Finally, activin A, β-cellulin, and nicotinamide were added. Activin A regulates the exogenesis of β-cells in vivo, while β-cellulin plays an important role in regulating growth and/or differentiation of pancreatic endocrine precursor cells (23). Nicotinamide is an effective inducer used to preserve islet viability through poly ADP-ribose polymerase (PARP) (21). In this initial study, exendin 4, glucagon-like peptide -1 (GLP-1), or other agents thought to increase differentiation in vivo were not added.
RT-PCR revealed distinct stage-specific gene expression patterns. At the end of differentiation, the profile of the transcription factors closely resembles that of human pancreatic islets. There is a temporal sequence of genes during activation for the development of α- and β-cells (17). Pdx1 is one of the well-studied transcription factors that are critical to both β-cell development and function (38). It controls the maturation of pancreatic islet cells by regulating insulin 1 and other downstream genes. It was shown that the expression of Pdx1 is switched on first, followed by other IPC-related genes (41). Ngn3 appears transiently during the development of the pancreas. It demarcates cells destined to form the endocrine pancreas (14). The transcription factor Rfx6 directs islet cell differentiation downstream of Ngn3 (37), and this remains at an elevated level similar to that in human islets. Pax4 is expressed in a subset of endocrine progenitors and is essential for the differentiation of α- and β-cell lineage (39). Evidence had been provided that MafB is expressed before MafA. It is suggested that MafB may have a dual role in the embryonic differentiation of both α- and β-cells while MafA may regulate replication/survival and function of β-cells after birth (28). MafA appears to mediate expression of other key islet β-cells genes, such as GLUT-2 and PC2 (25). The prohormone convertases PC1 and PC2 are endo-peptidases found in islets that convert the prohormones to mature hormones (40). Both convertases are found in adult β-cells, but only PC2 is found in adult β-cells where it converts proglucagon to glucagon (11).
Several investigators had argued that the presence of insulin in such cells does not indicate intrinsic insulin production. They suggest that insulin from the utilized culture media can be absorbed by and sequestrated in these cells (15, 26, 32). Immunofluorescence staining was positive for insulin, c-peptide, and glucagon with coexpression of insulin and c-peptide by the same cells confirming that proinsulin synthesis was occurring in the cells and not being derived from any insulin in the culture media. Moreover, the c-peptide-positive cells were not diving (Ki67 negative) and nonapoptotic since they were TUNEL negative. Localization of c-peptide immunoreactivity in the rough endoplasmic reticulum, as revealed by immunogold staining, provides an additional evidence of intrinsic synthesis of insulin.
The number of differentiated cells under the present condition was modest. Microscopical examination suggests that ~5% of cells were insulin positive. Insulin content of the differentiated cells was also modest, in comparison with equivalent protein levels in pancreatic islet cells. These secretory data are consistent with qPCR data, suggesting that insulin gene expression in the cellular fraction was <1% of that of human islets. This is likely to reflect not only the small number of differentiated cells in the cellular fraction but also a low insulin content of each cell in comparison with human or rodent β-cells. Evidence of glucose-sensitive insulin secretion suggests that the cellular machinery for glucose sensing is in place and that insulin release could be regulated by similar mechanisms as found in islet β-cells (34). Further studies are required to improve the yield of differentiated functioning cells.
This is the first study to compare cells from diabetic and nondiabetic donors. Long-term exposure of MSCs to high glucose concentrations in vivo could influence their sensitivity for growth and differentiation. There were no significant differences in the characteristics of growth, differentiation, or gene profiles of the cells obtained from different diabetic and nondiabetic donors. However, the insulin content of cells obtained from nondiabetic donors was significantly less than that of diabetic donors. It is unclear if this represents a genetic or environmental difference embedded in the stem cells or a variation as a result of the small number of donors examined in this study. Increasing the numbers of donors will allow further examination of this point. The relative expressions of different genes in diabetic and nondiabetic human donors at the end of differentiation remain to be determined.
Although the insulin content of the cells was small, transplantation of a large number of cells (1 million/ mouse) was capable of correcting hyperglycemia in vivo. One may also speculate that additional number of cells could have completed their differentiation in vivo. This was associated with the appearance of human insulin and c-peptide in the blood of these transplanted animals associated with a reduction of mouse insulin in the STZ-induced diabetes. No evidence was found to suggest regrowth of the islet β-cells in the mouse pancreas, which would have been reflected by an increase in mouse insulin in the blood samples. Although the profiles of the glucose tolerance tests of the transplanted mice were slightly higher than controls, they demonstrated a glucose-sensitive release of human insulin. On the other hand, mice treated with culture media or undifferentiated MSCs remained hyperglycemic and died after few days. All these findings strongly suggest that the donor cells had a central role in controlling blood glucose levels.
Taken together, all these experimental findings support the potential of adult human bone marrow-derived MSCs to differentiate into IPCs (3). Differentiation of autologous MSCs into IPCs would provide potential clinical benefits for diabetic patients. The donor could receive his or her own cells after differentiation, and although immunomodulation would be required for type 1 diabetic subjects, no suppression would be needed for type 2 diabetic subjects. The challenge is to demonstrate that enough insulin-positive cells with adequate functional capacity can be produced in culture without the requirement for molecular biological engineering. In addition, the duration during which these cells can maintain their active function in vivo, the needed numbers, and the optimal site for their implantation have also to be studied.
Footnotes
Acknowledgments
The study was funded by Misr El-Kheir Foundation (a charity nonprofit organization), Egypt. The efforts of Dr. Anne Clark from the Oxford Center for Diabetes, Endocrinology, and Metabolism in providing the confocal images and revising the manuscript are deeply appreciated. Electron microscopy nanogold processing was supported by Institutional Core Grant #CA 16672 High-Resolution Electron Microscopy Facility, UTMDACC. We thank Kenneth Dunner, Jr. for his excellent EM work. The authors declare no conflicts of interest.