
Abstract
Bone marrow mesenchymal stem cell (BM-MSC) transplantation has been suggested to be a promising method for the treatment of pulmonary arterial hypertension (PAH), a fatal disease currently without effective preventive/therapeutic strategies. However, the detailed mechanisms underlying BM-MSC therapy are largely unknown. We designed the present study to test the hypothesis that circulating platelets facilitate BM-MSC homing to the lung vasculature in a rat model of PAH induced by monocrotalin (MCT). A single subcutaneous administration of MCT induced a marked rise in right ventricular systolic pressure (RVSP) and the weight ratio of right to left ventricle plus septum (RV/LV+S) 3 weeks after injection. The injection of MSCs via tail vein 3 days after MCT significantly reduced the increase of RVSP and RV/LV+S. The fluorescence-labeled MSCs injected into the PAH rat circulation were found mostly distributed in the lungs, particularly on the pulmonary vascular wall, whereas cell homing was abolished by an anti-P-selectin antibody and the GPIIb/IIIa inhibitor tirofiban. Furthermore, using an in vitro flow chamber, we demonstrated that MSC adhesion to the major extracellular matrix collagen was facilitated by platelets and their P-selectin and GPIIb/IIIa. Therefore, the current study suggested that platelet-mediated MSC homing prevented the aggravation of MCT-induced rat PAH, via P-selectin and GPIIb/IIIa-mediated mechanisms.
Introduction
Pulmonary arterial hypertension (PAH) is an incurable disease characterized by high pulmonary arterial resistance and endothelial proliferation. The pathological features of the disease are medial hypertrophy and plexiform lesions, leading to vasoconstriction, vascular-wall remodeling, and in situ thrombosis (15,33). Although routine drug administration, such as vasodilators (e.g., prostacyclin), inhalation of nitric oxide, and endothelin receptor antagonists, has a modest impact on pulmonary hemodynamics, the effects do not persist (8,28). In spite of notable advances in the therapy of PAH during recent years, the curative effect and prognosis of this disease remains unpromising. Thus, new approaches that can protect or reverse vascular remodeling in the development of PAH are urgently needed.
Recently, mesenchymal stem cells (MSCs) (17,18, 26,41) and endothelial progenitor cells (EPCs) (39,42, 43) have been reported to be beneficial for treating cardiovascular diseases, especially PAH, both in animal models and human patients. Despite the significant experimental and clinical efficiency, the detailed mechanisms of cell therapy are poorly understood. Homing and engraftment are prerequisites for stem cells to display their activities in the target organ, especially when they are infused via the circulation. These complex processes depend on a temporal and spatial interplay between chemokines, chemokine receptors, intracellular signaling, adhesion molecules, and proteases (5). When vascular endothelium is injured, the coagulation system and the activation of platelets start simultaneously (7,24). We have also demonstrated an enhanced ex vivo platelet activation and platelet–leukocyte interaction in the rat model of PAH (14). Many studies have shown that activated platelets promote initial tethering, firm adhesion, migration, and differentiation of EPCs (6,21). Thus, we hypothesized that platelets play an important role in MSC adhesion to the injured lung, ultimately assisting them to repair or protect against the development of PAH.
In the present study, we demonstrated that the administration of MSCs significantly ameliorated monocrotalin (MCT)-induced PAH and right ventricle (RV) hypertrophy in rats and prolonged their survival time. More importantly, we found that platelet P-selectin and glycoprotein IIb/IIIa (GPIIb/IIIa; integrin αIIbβ3) played a key role in mediating MSC homing in vitro and in vivo.
Materials and Methods
Cell Isolation and Culture
BM-MSCs were isolated from the bone marrow of male Sprague-Dawley rats (Zhejiang University, China) as described previously (2). Briefly, 1-month-old rats were sacrificed in a CO2 chamber. Tibias and femurs were dissected, the ends of the bones were cut, and 10 ml DMEM (Gibco, Grand Island, NY) was injected into the central canal of the bone to extrude the marrow. After Histo-Paque purification (density 1.077 g/ml; Sigma, St. Louis, MO) of whole bone marrow, mononuclear cells were isolated and cultured at a density of 2 × 105 cells/cm2 in DMEM supplemented with 20% fetal bovine serum (Gibco) and 1% penicillin/streptomycin/amphotericin (Gibco). After 24 h, nonadherent cells were removed, and the medium was changed every 3–4 days until the culture became 80% confluent. The cells were trypsinized, split 1:4, and passaged up to three times. Differentiation of MSCs into adipocytes, osteocytes, and chondrocytes was described previously (31).
Flow Cytometry
Cultured BM-MSCs were analyzed in an FC-500 flow cytometer (Beckman Coulter, Fullerton, CA) by using a fluorescein isothiocyanate-conjugated mouse monoclonal antibody against rat CD44 (clone OX-49; BD Biosciences, Franklin Lakes, NJ) and phycoerythrinconjugated mouse monoclonal antibodies against rat CD90 (sc-19614; Santa Cruz Biotechnology Inc., Santa Cruz, CA), CD45 (clone OX-1; eBioscience, San Diego, CA), and CD29 (clone HMb1-1; eBioscience). Isotypic antibodies served as controls.
Experimental Protocol of MCT-Induced PAH in Rats
All protocols were maintained in accordance with the Institutional Animal Care and Use Committee of Zhejiang University.
Sprague-Dawley rats weighing between 180 and 250 g received a single subcutaneous injection of 60 mg/kg MCT (Sigma-Aldrich, St Louis, MO). Three days after MCT injection, rats were randomly assigned to two groups: rats that received no treatment (MCT group, n = 8) and rats that received 1 ml (4 × 106) MSCs (MCT + MSCs group, n = 43). For the negative control, rats received a subcutaneous injection of 0.9% saline instead of MCT (control group, n = 18). Cells were delivered via tail vein 3 days after MCT and rats were euthanized with CO2 at 21 days. Twenty-one days after MCT injection, right ventricular systolic pressure (RVSP) and the ratio of right to left ventricular plus septal weight (RV/LV+S) were determined as described previously (4,27).
Lung Vascular Morphometry
The lung was fixed by vascular perfusion through the right ventricle as described previously (23). Briefly, the rat was anesthetized with an intraperitoneal injection of 4% chloral hydrate, and a needle was inserted into the right ventricle while incising the left atrium at the same time. This was followed by a 37°C 0.9% saline perfusion. When the outflow appeared clear of red blood cells, this was changed to ice-cold 4% paraformaldehyde (200–300 ml). The lung was removed and fixed in 4% formaldehyde for another 2 h at 4°C. Lung tissue sections were prepared and stained with hematoxylin and eosin as described previously (11,12). The percent medial wall thickness of each artery was calculated with the following formula as previously described (9): index (%) = (external diameter - internal diameter)/external diameter x 100. For each rat, 10–20 vessels were counted and the average was calculated (n = 6 for each group).
Platelet Preparation
To prepare washed platelets, blood was centrifuged at 150 × g for 20 min, and the platelet-rich plasma (PRP) was collected. The PRP was centrifuged for 10 min at 800 × g and the platelet pellet was then resuspended in Tyrode buffer at the concentration of 2.5 × 108 platelets/ml.
Flow Chamber Technique
Glass coverslips (50 × 24 mm, Eupotubo, Amadora, Portugal) were coated with 100 μl 0.08 μg/cm2 collagen (Chrono-Log Corp, Havertown, PA) overnight at 4°C.
Static Conditions
The interaction was measured by a regular adhesion assay (21). The slides were rinsed with PBS to remove unbound collagen and then incubated with calcein red–orange (Invitrogen, Carlsbad, CA)-labeled washed platelets for 10 min at room temperature to form a monolayer of activated platelets. Afterwards, a calcein green AM (Invitrogen)-labeled MSC suspension (4 × 106/ml) was incubated on collagen-coated slides in the absence or presence of a platelet monolayer for 15 min at room temperature. Finally, the slides were rinsed with PBS to remove unbound cells. MSCs on the slides were directly visualized under a Nikon TE2000-U intravital fluorescence microscope equipped with a Nikon DS-2MBWc-U1 CCD video camera (Nikon Corp., Tokyo, Japan). The total numbers of adherent MSCs were counted in 10 randomly selected fields (magnification 100x).
Flow Conditions
The coverslip was assembled in a parallel-plate perfusion chamber equipped with a peristaltic tubing pump (Masterflex L/S, Cole-Parmer, USA) (36) and the chamber was mounted on the object platform of the fluorescence microscope. The tubing-flow chamber system was then filled with Tyrode-HEPES buffer containing 1% BSA and allowed to equilibrate for 10 min at room temperature. The coverslip was then perfused with a calcein red–orange-labeled MSC suspension at a shear rate of 1,000 s−1 in the absence or presence of calcein green AM-labeled platelets. Ten minutes after perfusion, the coverslip was perfused with PBS for 2 min to remove nonadherent cells. The images from at least 10 separate fields per flow experiment were recorded.
Adhesion assays were also used to test the effect of P-selectin monoclonal antibody (10 μg/ml) (clone 9E1; R&D Systems Ltd., Abingdon, UK) and tirofiban (15 Hg/ml) (Wuhan Grand Pharmaceutical Group, Wuhan, China). All images of platelet deposition and MSC adhesion were illuminated at 450–490 nm and 510–560 nm excitation. The images were analyzed by Image-Pro Plus version 5.3 (MediaCybernetics Inc., Bethesda, MD).
Scanning Electron Microscopy (SEM)
Glass coverslips were coated with collagen and covered with a monolayer of platelets. Then MSCs were seeded onto slides and rinsed with PBS. Two hours after seeding, the cell morphology and distribution were visualized under a scanning electron microscope. The specimens were fixed with 0.25% glutaraldehyde for 4 h and rinsed three times with PBS; they were then immersed in OsO4 for 1 h, rinsed three times with PBS, and dehydrated in increasing concentrations of acetone (30–100% v/v). After drying, the specimens were mounted on aluminum stubs and coated with gold, then viewed in a Hitachi S-3000N SEM (Hitachi, Japan) at an accelerating voltage of 20–25 kV.
Fluorescent Labeling and Engraftment of Cells
Before transplantation, 4 × 106 cells were incubated for 20 min with 3 μl carboxyfluorescein diacetate (CFDA; Invitrogen) and injected via tail vein at 3 days after MCT injection. Lungs were perfused and fixed with 4% paraformaldehyde before harvesting at different time points after cell delivery. After OCT embedding, 10-μm-thick lung frozen sections were prepared. DAPI was used for nuclear staining and images were captured by a Zeiss confocal fluorescence microscope (Zeiss LSM510 Meta, Oberkochen, Germany).
Analysis of Rat Tissues for Mesenchymal Stem Cell Homing
Thirty minutes after the administration of vehicle, platelet P-selectin blocking antibody 9E1 (10 μg/kg bolus), or the GPIIb/IIIa inhibitor tirofiban (200 μg/kg bolus) via tail vein, CFDA (Invitrogen)-labeled cell transplantation was performed. At designated time points (7, 14, and 21 days), cardiac perfusion was performed with 37°C 0.9% saline to remove nonadhered MSCs. Afterwards, the lungs and spleen were removed. Single-cell suspensions were made as previously described (17). Briefly, tissues were gently eased through a 200-mesh filter, treated with erythrocyte lysis buffer (in mM: 155 NH4Cl, 20 NaHCO3, 1 EDTA) for 4 min on ice, and washed twice in medium with 5% FCS. Samples were analyzed on an FC-500 flow cytometer, and at least 100,000 events were acquired and analyzed using CXP Analysis Software Version 2.1. Homing results were expressed as the percentage and the number of CFDA-positive cells per 100,000 events.
Statistical Analysis
Data are presented as mean ± SEM using SPSS v16.0. Differences between groups were evaluated by ANOVA, and by Tukey's multiple-group comparisons test. Differences within groups between the lung and spleen were assessed using a paired t-test. The survival data are presented as Kaplan-Meier curves and compared by log-rank test. A value of p < 0.05 was considered statistically significant.
Results
Characterization of BM-MSCs
Most cells were spindle shaped (Fig. 1A). To evaluate the multipotency of BM-MSCs, we induced cell differentiation into adipocytes, osteocytes, and chondrocytes. Adipogenic differentiation (Fig. 1B) was induced in expanded MSC cultures with 1-methyl-3-isobutylxanthine, dexamethasone, insulin, and indomethacin. Osteogenic differentiation (Fig. 1C) was induced by glycerol phosphate, dexamethasone, and ascorbic acid. Chondrogenic differentiation (Fig. 1D) was induced in MSC mass cultures with transforming growth factor-β3 (TGF-β3). Positive induction was detected by von Kossa, safranin O, and oil red staining. Flow cytometric analysis of cultured BM-MSCs demonstrated that they were positive for CD90 (98.3%), CD29 (99.0%), and CD44 (96.5%), but negative for CD45 (2.58%) (Fig. 1E).
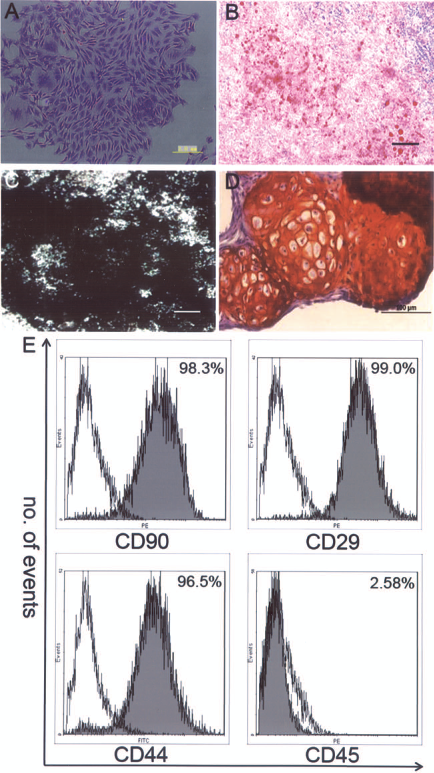
Characterization of bone marrow rat mesenchymal stem cells (BM-MSCs). (A) Morphology of BM-MSCs. (B–D) Multipotency of BM-MSCs: differentiation into (B) adipocytes, (C) osteocytes, and (D) chondrocytes. (E) Flow cytometric analysis of BM-MSC surface receptors. Gray areas indicate staining with a specific antibody (CD90, CD29, CD44, and CD45) and unfilled areas represent staining with isotypic control antibodies. Representative results from one of three individual experiments are shown. Scale bars: 2 mm (A), 50 μm (B, C), and 100 μm (D).
Stable Adhesion of Fluorescence-Labeled MSCs on the Arterioles of the Lung
CFDA-labeled MSCs were injected via tail vein 3 days after administration of MCT. Seven days after delivery, fluorescence-tagged MSCs were seen stably adhered on the arterioles of the lung (Fig. 2).
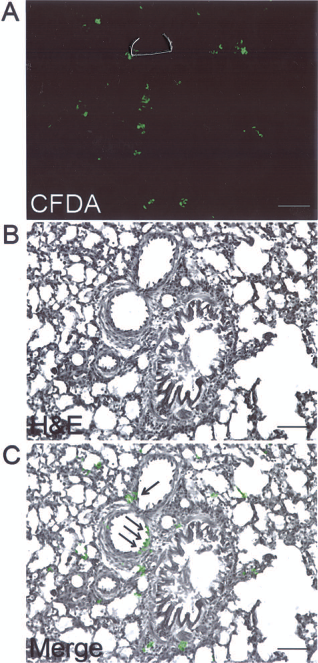
Microscopic analysis of transplanted CFDA-labeled MSCs. MSCs were transplanted via tail vein 3 days after MCT injection. (A) Carboxyfluorescein diacetate (CFDA)-positive (green) MSCs. (B) H&E stain. (C) Merge of (A) and (B). The arrows indicate MSCs incorporated into a pulmonary vessel. Scale bars: 200 μm.
Effect of Early Injection of MSCs on RVSP, RV/LV+S, and Survival
RVSP was significantly increased at 21 days after MCT treatment, compared with saline-treated controls (40.20 ± 3.46 vs. 21.00 ± 0.55 mmHg) (Fig. 3A). Right ventricular hypertrophy, as measured by the RV/LV+S ratio, was increased in MCT rats (0.35 ± 0.03 vs. 0.22 ± 0.01 control) (Fig. 3B). The delivery of MSCs significantly prevented the rise in pulmonary systolic pressure (28.84 ± 1.35 mmHg, p < 0.05 vs. MCT) (Fig. 3A) and reduced right ventricular hypertrophy (0.26 ± 0.02, p < 0.05 vs. MCT) (Fig. 3B) at 21 days after MCT. However, the levels of RVSP and RV/LV+S in the MSC group were still higher than those of the control group. Moreover, it did not rescue the animals when the MSCs were injected 3 weeks after MCT, when clear symptoms started to emerge (RVSP 35.75 ± 0.87 mmHg; RV/LV+S 0.33 ± 0.02; p > 0.05 vs. MCT for both). Furthermore, in survival analysis, the injection of MSCs mildly reduced MCT-induced mortality (Fig. 3C).

Hemodynamics and survival rate analysis. Right ventricular systolic pressure (RVSP) (A) and RV/LV + septum weight ratio (B) were measured in the indicated groups. Survival rate of rats in the indicated groups are presented in the Kaplan-Meier curves (C). *p < 0.0001, **p < 0.05 when indicated groups are compared (n = 8–10 for each group). MCT, monocrotalin; MSCs, mesenchymal stem cells.
Pulmonary Artery Remodeling
Three weeks after MCT injection, the lungs were full of microthrombi and ischemic foci, whereas in MSC-treated rats the appearance of the lung was similar to control rats (Fig. 4A). Pulmonary arteries of MCT rats showed an increased medial wall thickness compared with controls (53.82 ± 2.97% vs. 16.63 ± 1.37%, p < 0.0001) (Fig. 4B) and MSC administration significantly reduced the medial wall thickness (36.24 ± 1.51%, p < 0.0001 vs. MCT) (Fig. 4B). In addition, injection of MSCs into control rats had no effect on the pulmonary artery medial wall thickness (data not shown).
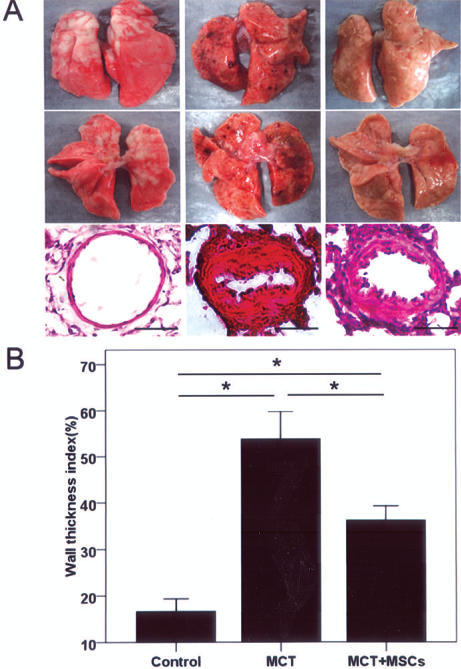
Effect of MSCs on vascular remodeling. Rats (n = 8–10) from each group were sacrificed 3 weeks after MCT injection to determine the effect of MSCs. Images represent at least three independent experiments. Lungs and H&E staining of lungs from indicated groups are shown in (A). Scale bars: 25 μm. (B) Quantification of pulmonary arterial wall thickness. *p < 0.0001 when indicated groups are compared.
Platelet–MSC Interaction Under Static Conditions
Cell adhesion assays were performed to investigate the platelet (PLT)–MSC interaction under static conditions and to validate the role of platelet P-selectin and GPIIb/IIIa (Fig. 5). Isolated MSCs were deposited on collagen-coated glass in the presence or absence of a monolayer of platelets. MSCs adhered to activated platelets (Fig. 5B), but not to the collagen surface alone (Fig. 5A) (44.25 ± 1.36 vs. 4.88 ± 1.59, p < 0.0001) (Fig. 5I). In addition, the MSC adhesion was blocked by a monoclonal antibody against P-selectin 9E1 or tirofiban, a platelet integrin GPIIb/IIIa inhibitor (14.07 ± 2.24 with 9E1, 15.44 ± 1.53 with tirofiban, p < 0.0001 vs. MSCs + PLTs) (Fig. 5I), indicating that the MSC–platelet interaction was mediated by platelet P-selectin and GPIIb/IIIa (Fig. 5C, D).

Interaction between platelets and MSCs under static and flow conditions. Representative images of static adhesion (A–D) and adhesion under flow at a shear rate of 1000 s−1 (E–H) on different adhesive surfaces. From left to right: collagen alone (A, E); collagen + platelet monolayer (B, F); collagen + platelets + 9E1 (C, G); and collagen + platelets + tirofiban (D, H). Magnification: 100x. MSCs were labeled with calcein red and platelets were labeled with calcein green. Quantification of adherent MSCs was calculated under static (I) and flow (J) conditions. *p < 0.0001 when indicated groups are compared. Scanning electron microscopy revealed: (K) collagen, (L) platelet aggregation, (M) interaction between MSCs and platelets; the frame in (M) is magnified in (N). PLTs, platelets. Scale bars: 50 μm (A–H), 3 μm (K, L), 10 μm (M), and 5 μm (N).
SEM was used to observe MSC–platelet interaction on collagen directly. Activated platelets formed thrombi on filamentous collagen (Fig. 5K–N). MSCs adhered to the surface of collagen-coated glass where activated platelets were present (Fig. 5M, N).
Platelet–MSC Interaction Under Flow Conditions
To test the capacity of platelets to support the adhesion of MSCs to collagen under flow conditions, reconstituted blood and labeled MSCs in the absence or presence of platelets were perfused over a collagen-coated surface at the shear rate of 1000 s−1, which corresponded to arterial shear rate (10). As expected, MSCs failed to adhere to the collagen surface without platelets (Fig. 5E). In contrast, MSCs rapidly tethered to areas with high platelet density and the tethered MSCs either adhered immediately to the platelets or rolled a short distance before becoming adherent a few seconds later (Fig. 5F). In line with the adhesion study under static conditions, anti-P-selectin antibody 9E1 and tirofiban both blocked MSC–platelet interaction in flow (4.73 ± 0.65 with 9E1, 3.86 ± 0.64 with tirofiban, vs. 50.12 ± 3.85 with MSC + PLTs, p < 0.0001) (Fig. 5G, H, J). These results further showed that platelet P-selectin and GPIIb/IIIa were involved in MSC adhesion.
In Vivo Homing of Mesenchymal Stem Cells in PAH Rats
CFDA-labeled rat MSCs (4.0 × 106 cells/animal) were administered via tail vein to animals 3 days after MCT treatment. Animals were sacrificed at indicated times and single-cell suspensions from lungs and spleen were analyzed for CFDA-positive cells by flow cytometry. Compared with control group, the level of homing was increased in lungs from MSC-injected rats after 7 days (Fig. 6A). However, the homing of MSCs to the spleen was milder, although it also increased after 21 days (Fig. 6A). With time, more and more CFDA-labeled cells engrafted into the lungs compared with the spleen and control rats over 21 days (Fig. 6A, B). The MSCs found in the lung 21 day after transplantation were estimated 0.25–0.375% of those originally administered. Furthermore, PAH rats treated with anti-P-selectin antibody 9E1 or tirofiban exhibited a significantly reduced level of cell homing to the lungs 7 days after cell injection, compared with control MSC-treated PAH rats (889.5 ± 12.96 with 9E1, 869.8 ± 40.48 with tirofiban, compared with 1824.0 ± 36.29 in control; p < 0.0001 for both) (Fig. 6B, C). Antiplatelet treatment by tirofiban or 9E1 also abrogated the protective effect of MSCs, as demonstrated by the increased RVSP in MCT model (Fig. 6D).
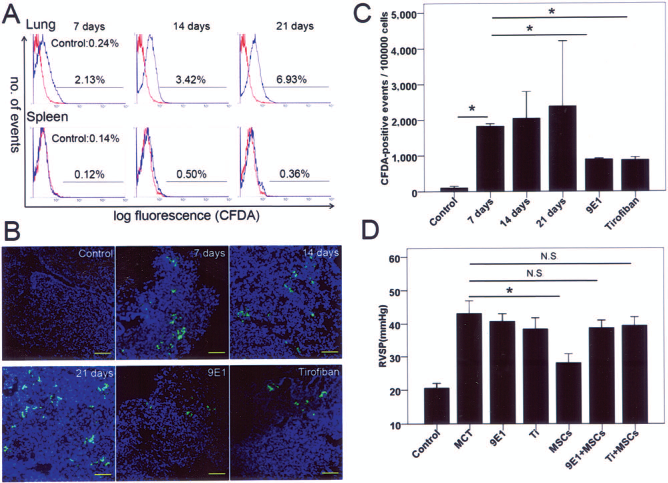
In vivo homing of rat mesenchymal stem cells. All animals were injected with MCT 3 days before CFDA-labeled MSC transplantation. (A) Representative flow cytometric analysis of cell suspensions from lung (upper panel) and spleen (lower panel) of rats (red: control, MCT rat infused with saline; blue: MCT rat infused with MSCs after 7, 14, 21 days, respectively). Bars indicate the percentage of CFDA-positive events. (B) Representative microscope images of explanted lung at different time points. Green: CFDA-positive MSCs; blue: DAPI. Scale bars: 100 μm. (C) Quantification of fluorescently labeled cells in lungs over a period of 3 weeks after cell transplantation. *p < 0.0001 when indicated groups are compared. (D) Right ventricular systolic pressure in the indicated groups. Ti, tirofiban. *p < 0.0001 when indicated groups are compared; N.S., no significance.
Discussion
In the current study, we have shown that the engraftment of bone marrow-derived mesenchymal stem cells resulted in the amelioration of MCT-induced rat PAH. We have also demonstrated that platelet P-selectin and GPIIb/IIIa were important in facilitating MSC adhesion to a collagen surface in vitro and MSC homing in vivo. This is, to our knowledge, the first study to investigate the role of platelets in MSC therapy in a PAH model.
The MCT-treated rat is a well-known model of PAH, characterized by injuries of the pulmonary arterial endothelium and medial thickening. In the present study, both pulmonary vascular resistance and pulmonary arterial pressure increased two- to threefold, accompanied by significant right heart hypertrophy, evidenced by the increased weight ratio of right to left ventricle plus septum. Pulmonary arterial remodeling was also evident, as shown histologically by the marked medial hypertrophy of these vessels.
In the present study, MSCs were chosen as the cell source for several reasons. First, large amounts of MSCs are easy to obtain, which is a great advantage in therapeutic practice. Second, MSCs have therapeutic implications in vascular diseases, due to their multipotent ability to differentiate into cell lineages including endothelial progenitor cells (16,29,32,37). Third, MSCs have favorable characteristics for the genetically modified cell seeding technique (13), which provides margins for the future improvement of this cell therapy strategy. In the present animal model of severe PAH, treatment with MSCs significantly attenuated PAH development, as evidenced by the improved hemodynamic values, recovery of both right ventricular hypertrophy and pulmonary vessel remodeling, and prolonged animal survival time, when compared to untreated PAH rats. However, the MSC-treated group still had higher levels of RVSP and RV/LV+S than those of the control group. Moreover, MSC transplantation did not rescue the animals when injected 3 weeks after MCT, when evident symptoms started to emerge. Our results indicate the importance of early intervention. In this model, the average time from the emergence of symptoms to death is 1–3 weeks. With such a limited therapeutic time window, the favorable effects of MSCs may not be sufficient to counteract the exacerbating PAH pathogenesis. Moreover, the rapid progression of PAH was associated with systemic inflammatory responses, which could negatively regulate the fate of MSCs in vivo by, for example, activated natural killer cells (35) or elevated concentrations of cytokines like tumor necrosis factor-α (TNF-α and interleukin-6 (IL-6) (40). The unfavorable microenvironment and possible MSC loss might also explain why even the early administration of MSCs only had limited effects on prolonging survival, with the mortality increasing suddenly from the fourth week after MCT treatment. As a limitation of the current study, we only followed the engrafted cells until 21 days. A longer follow-up, more detailed cell fate analysis, and a repeated MSC administration approach are thus warranted in future studies.
Our in vitro study demonstrated the interaction between MSCs and activated platelets under both static and flow conditions. The platelet–MSC interaction appeared to be dependent on platelet P-selectin. It has to be noted that the adhesion molecule CD44 is highly expressed on MSCs, as measured by flow cytometry in the present study. CD44 has been reported as a ligand of P-selectin and can promote MSC adhesion to extracellular matrix (44). It is thus possible that CD44–P-selectin is one of the major molecular pairs supporting platelet–MSC interaction. The present study also demonstrated that platelet GPIIb/IIIa may play important roles in platelet–MSC interaction. Tirofiban, an inhibitor targeted against activated high-affinity GPIIb/IIIa, significantly inhibited MSC adhesion to platelets. Thus, platelet–MSC interaction may also be facilitated by the concerted action of platelet integrin GPIIb/IIIa, fibronectin (38), and α4 and β1 integrins (very late antigen 4; VLA-4) (34) on MSCs. In addition, chemokines expressed by platelets such as stromal cell-derived factor-1 (SDF-1) may also play roles in the recruitment of MSCs (17,20,25), and inhibition of the SDF-1/CXCR4 (C-X-C chemokine receptor type 4) pathway may partially block cell homing (1).
To further confirm that MSC engraftment is dependent on platelets, a previously described method was used to quantify the distribution of MSCs in vivo after their injection into the circulation (19). We found that transplanted cells were capable of persistently localizing to the MCT-injured lungs, but less to other organs such as the spleen. It has to be noted that MSCs seemed to be “trapped” instead of firmly adhered to the vasculature during the early time (before 7 days) after their transplantation. This was obvious when the perfusion process washed away the nonadherent cells in the lungs. Thus, homing of MSCs was only evident 1 week after cell transplantation. Interestingly, this is in agreement with the platelet activation kinetics in this PAH rat model; as we have previously shown, clear platelet activation only starts to emerge at least 1 week after MCT injection (14). Thus, it appears that MSC homing is correlated with platelet activation in vivo. The homing dependence on platelet activation may also partly explain the failure to rescue animals when MSCs were used after the emergence of symptoms. In late-stage PAH, platelets are more activated and tend to form thrombi and attach to leukocytes, thus reducing their propensity to interact with engrafted MSCs. Importantly, in vivo blockade with tirofiban and a P-selectin antibody indicated that the homing efficiency of MSCs was critically dependent on platelet P-selectin and GPIIb/IIIa, which was consistent with the in vitro study. Moreover, this platelet-dependent cell homing seems important in the protective effect of MSCs in vivo, as MSCs prevented RVSP increase was abrogated by antiplatelet treatment. Platelet and MSC interaction may have similarities to the interaction between platelets and leukocytes at the site of vascular injury. Under physiologic flow conditions, leukocytes tether to and roll on adherent activated platelets in a P-selectin- and GPIIb/IIIa-dependent manner (3,22). As previously suggested (25), either MSCs or EPCs might have adhesion behavior and dependence on activated platelets similar to those of leukocytes. Thus, upon vascular injury, activated platelets first tether and adhere to exposed subendothelium, when expressed P-selectin and activated high-affinity GPIIb/IIIa may subsequently facilitate MSC recruitment to injured vessels (Fig. 7). The present observations indicate that it is important to take activated platelets into consideration in MSC-based therapies to treat vascular diseases. Yet it is also important to keep in mind that platelet activation plays pivotal roles in the pathogenesis of PAH (30). Platelets are therefore a double-edged sword; the intricate regulation of their activation status to minimize their influence on thrombosis and inflammation, and to amplify the functions of promoting MSC homing, may finally turn the sword into a plowshare.
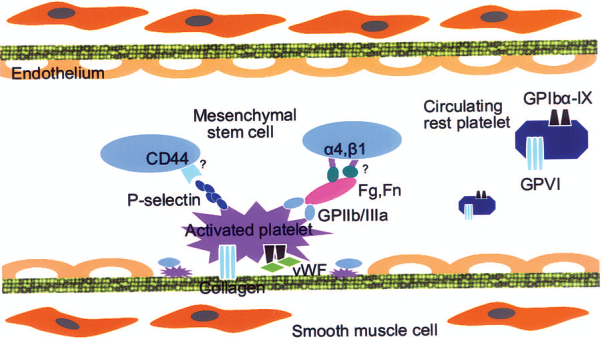
Schematic diagram of activated platelet recruitment of BM-MSCs to injured vessel wall. Upon vessel wall injury, endothelial cells are damaged and platelets adhere to the exposed collagen in a process involving glycoprotein VI (GPVI) and GPIbα-IX. P-selectin and activated GPIIb/IIIa are expressed on the activated platelet surface, thereby facilitating MSC recruitment, presumably via interaction with CD44 and integrin α4 and β1 on MSCs. Fg, fibrinogen; Fn, fibrinectin; vWF, von Willebrand factor.
Taken together, the administration of MSCs ameliorated, but did not cure, MCT-induced PAH. The in vivo homing of MSCs was dependent on platelet activation, and subsequent P-selectin expression and GPIIb/IIIa activation. Our findings may have implications in optimizing cell-based strategies for the treatment of severe PAH patients.
Footnotes
Acknowledgments
We thank Prof. Iain Bruce for critical reading of the manuscript. This work was supported by research grants from the National Natural Science Foundation of China (30970656), National Basic Research Program of China (2012CB966603), the Science Foundation of Chinese Universities, the Scientific Research Foundation for Returned Overseas Chinese Scholars, the Education Ministry and State Personnel Ministry of China, and the Swedish Research Council. The authors declare no conflicts of interest.