
Abstract
Neurotrophic factors (NTFs) are involved in the regulation of neuronal survival and function and, thus, may be used to treat neurological diseases associated with neuronal death. A major hurdle for their clinical application is the delivery mode. We describe here a new strategy based on the use of progenitor cells called mesoangioblasts (MABs). MABs can be isolated from postnatal mesoderm tissues and, because of a high adhesin-dependent migratory capacity, can reach perivascular targets especially in damaged areas. We generated genetically modified MABs producing nerve growth factor (MABs-NGF) or brain-derived neurotrophic factor (MABs-BDNF) and assessed their bystander effects in vitro using PC12 cells, primary cultures, and organotypic cultures of adult hippocampal slices. MABs-NGF-conditioned medium induced differentiation of PC12 cells, while MABs-BDNF-conditioned medium increased viability of cultured neurons and slices. Slices cultured with MABs-BDNF medium also better retained their morphology and functional connections, and all these effects were abolished by the TrkB kinase blocker K252a or the BDNF scavenger TrkB-IgG. Interestingly, the amount of BDNF released by MABs-BDNF produced greater effects than an identical amount of recombinant BDNF, suggesting that other NTFs produced by MABs synergize with BDNF. Thus, MABs can be an effective vehicle for NTF delivery, promoting differentiation, survival, and functionality of neurons. In summary, MABs hold distinct advantages over other currently evaluated approaches for NTF delivery in the CNS, including synergy of MAB-produced NTF with the neurotrophins. Since MABs may be capable of homing into damaged brain areas, they represent a conceptually novel, promising therapeutic approach to treat neurodegenerative diseases.
Introduction
Neurotrophic factors (NTFs) are critical for the survival, development, and function of neurons in the mammalian central nervous system (23). Alterations in NTFs or their receptors leading to loss of function can cause neuronal death and contribute to the pathogenesis of neurodegenerative diseases and of diseases associated with neuronal damage (4,7,11,67). Not surprisingly, therefore, they received considerable interest as therapeutic agents (5,56,60).
A particular attention has been devoted to the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF). A vast literature shows that NGF and BDNF exert trophic and neuroprotective effects on neurons, but their clinical use is hindered by a short biological half-life, a poor blood–brain barrier (BBB) permeability, and side effects due to their pleiotropic systemic actions (2). Given these difficulties, the attention turned into the development of techniques that allow local delivery based on gene therapy (49,61) or on cell-mediated approaches.
Stem cell-mediated gene delivery, by which stem cells are genetically modified with NTFs and then transplanted into the brain, is emerging as a promising strategy. This strategy has been already validated for several lines of stem cells, including neural (26,39,42), embryonic (34,40,52), and mesenchymal stem cells (20,36,54): when engineered to produce NTFs, these cells were found to ameliorate experimental neurodegenerative diseases or injuries. The advantages of using genetically modified stem cells as grafts include the fact that they could exert not only long-term functional integration but also repair capabilities and that they can ensure a continuous and concentrated local supplementation of diffusible therapeutic molecules (like NGF or BDNF), reducing nonselective delivery, and allowing high treatment efficiencies for long time periods. However, the downside of these approaches is that they require an invasive delivery route to the target location in the nervous system, a problem that severely limits their potential for human therapy.
These considerations prompt the search for alternative cell sources that display more favorable homing properties, avoiding the necessity of a direct injection into the brain. Here, we explored the feasibility of a new approach: the use of multipotent, mesodermal progenitor cells [mesoangioblasts (MABs)] that constitutively produce a subset of NTFs [e.g., vascular endothelial growth factor B (VEGF B), fibroblast growth factor-2 (FGF-2), FGF-7, platelet-derived growth factor AA (PDGF)] and can be engineered to produce others. MABs are an affordable cellular source since they can be isolated from small biopsies of postnatal human skeletal muscle and have already been utilized for regenerative purposes in large animal models, besides rodents (47,53). More importantly, a phase I/phase II cell therapy trial with donor MABs for patients affected by Duchenne muscular dystrophy is planned for year 2011 (59).
MABs have a high, adhesin-dependent, migratory capacity and the ability to cross the vessel wall and reach perivascular targets, a property not demonstrated so far for mesenchymal stem cells. Their chemotactic ability is increased under inflammatory conditions, when the adhesin–integrin system and diapedesis moving are activated (16). When peripherally administered, therefore, MABs may selectively cross the BBB and home into lesioned brain parenchyma. These cells express several neuro-ectodermal genes, but they do not differentiate into neurons (58). Thus, they may not be used for a restorative engraftment. Instead, MABs are expected to act as a reservoir and delivery system for NTFs.
The aim of this study was to engineer MABs to produce and secrete NGF or BDNF and to examine their bystander effects. Thus, the medium collected from MAB cultures (containing either NGF or BDNF plus other constitutively produced NTFs) has been applied to PC12 cells, primary neuronal cultures (PC), and organotypic cultures (OC) of adult hippocampal slices.
Materials and Methods
Expression Vectors
Murine preproNGF and rat preproBDNF cDNAs have been PCR amplified from pTrcHisA-preproNGF and pBK-CMV-preproBDNF using the following XhoI-5′and NotI-3′ primers: 5′ primer-GTCGACCTCGAGGTAATGTCCAT GTTGTTCTACACTCTG and 3′ primer-AGTCGGCTG-C GGCCGCTCAGCCTCTTCTTGTAGCCTTCCT for NGF; 5′ primer-GTCGACCTCGAGAT-GACCATCCTTTTC CTTACTATG and 3′ primer-AGTCGGCTGCGGCCGC CTATCTTCCCC-TTTTAATGGTCAG for BDNF. The PCR reaction has been performed with proofreading Pfu DNA polymerase (Promega, WI, USA) using the following conditions: 95°C, 1 min; 55°C, 1 min; 72°C, 1 min, for 25 cycles. Amplified preproNGF and preproBDNF cDNAs have been sequenced with the CEQ DTCS Quick Start Kit (Beckman Coulter, Inc., CA, USA) and then Xho/NotI cloned into the single chain variable fragment (ScFv) express eukaryotic vector (50) to produce ScFv-preproNGF and ScFv-preproBDNF constructs, respectively.
Transfection of Cells and Neurotrophin Quantification
COS cells (5 × 105; from the American Type Culture Collection, ATCC), grown for 1 day in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS), were transiently transfected with ScFv-preproNGF or ScFv-preproBDNF using Fugene, according to the manufacturer's instructions (Roche, Basel, CH). NGF and BDNF expression and secretion were evaluated on the conditioned medium 72 h after transfection.
Green fluorescent protein (GFP)-expressing MABs (45), grown in DMEM plus 10% FBS, have been stably transfected with the expression vectors ScFv-preproNGF or ScFv-preproBDNF using Lipofectamine (Invitrogen, CA, USA), according to manufacturer's instructions. Upon G418 selection, single clones of stable transfectants have been cultured with the ordinary methods, using the following culture medium (here referred to as Medium-1): 88% DMEM, 10% FBS, 0.5% GlutaMaxII, 22 mM mg/ml glucose, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 mg/ml streptomycin (all from Gibco, Invitrogen, CA, USA). The production of exogenous neurotrophins was analyzed by a “two-site sandwich” ELISA (8), performed on the medium collected from NGF- and BDNF-producing MABs. The medium was collected and centrifuged for 5 min at 230 x g, and then the supernatant was collected. The supernatant consists of the medium containing the soluble substances secreted by MABs, including NGF in the case of MABs-NGF and BDNF in the case of MABs-BDNF. NGF and BDNF “coating” were performed using anti-NGF mAb αD11 (1 ng/ml) and IgG-TrkB immunoadhesin (5 ng/ml), respectively. The following antibodies were used for ELISA: rabbit polyclonal anti-mNGF (30 μg/ml, Sigma, St. Louis, MO, USA); chick polyclonal anti-hBDNF (1 μg/ml, Promega, WI, USA); biotinylated anti-rabbit secondary antibody (1:1,000, Vector Laboratories, Inc., CA, USA); biotinylated anti-chick secondary antibody (1:1,000, Promega, WI, USA). As standards, recombinant 2.5S mNGF and hBDNF (Alomone Labs Ltd., Jerusalem, Israel) were used. Immunoreactivity was visualized with the ABC kit (Vector Laboratories) using tetramethylbenzidine (TMB) as substrate. Data are reported as average of at least two independent experiments, each performed in duplicate.
Western Blot Analysis
MABs-NGF (clone F10) and MABs-BDNF (clone A9) were compared with control MABs. The conditioned media were collected from cells grown for 3 days. Samples of conditioned media were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 10% acrylamide), and proteins were transferred onto Hybond-C nitrocellulose in transfer buffer (25 mM Tris, 192 mM glycine, 20% v/v methanol) for 1 h at 50 mA. The membrane was blocked for 12 h at 4°C in 5% milk–PBS (w/v). The following antibodies were used: rabbit polyclonal anti-murine NGF (30 μg/ml, 1:1,000; Sigma); rabbit polyclonal anti-BDNF (1:600; Santa Cruz Biotechnology); horse radish peroxidase (HRP) anti-rabbit (1:7,000 for NGF, 1:3,000 for BDNF) as secondary antibody. For NGF, the primary antibody was diluted in PBS–5% milk and incubated 1 h at room temperature; for BDNF, it was diluted in PBS–2.5% milk–1% bovine serum albumin (BSA) and incubated overnight at 4°C. Washes were performed in PBS–0.05% Tween and then PBS 1x. Membranes were developed using the ABC (Vector Laboratories) followed by the Amersham ECL kit (GE Healthcare, Little Chalfont, UK).
Assessment of NGF Biological Activity
PC12 Cells
PC12 cells (ATCC) (19) have been cultured in RPMI (Invitrogen, CA, USA) plus 10% FBS. Upon washing in serum-free medium, 4×105 PC12 cells were plated in 35 mm collagen-treated Petri dishes in the following different conditions: alone, in coculture with the same amount of MABs-NGF, in the presence of a 5-day-culture supernatant from MABs and MABs-NGF, or in the presence of recombinant NGF (50 ng/ml). PC12 cells were maintained in culture for 7 days, replacing fresh medium every 2 days. Neuronal differentiation was evaluated by the presence of neurite processes in the cultures and confirmed by β-tubulin immunofluorescence.
For β-tubulin and GFP expression in PC12/MABs coculture, cells were fixed in 4% paraformaldehyde (PFA), washed briefly in phosphate-buffered solution (PBS), and permeabilized with 0.1% Triton in PBS. After blocking in 10% normal goat serum (NGS), cells were stained with mouse anti-β III tubulin antibody TuJ1 (1:250, Covance, NJ, USA) and rabbit anti-GFP antibody (1:500; Invitrogen, CA, USA), followed by Alexa Fluor 594-conjugated anti-mouse and Alexa Fluor 488-conjugated anti-rabbit antibodies (1:500; Invitrogen, CA, USA).
Assessment of BDNF Biological Activity
Primary Hippocampal Neuronal Cultures
Primary hippocampal neuronal cultures (PCs) were derived from P0 newborn Swiss mice (Harlan Italy, San Pietro al Natisone, Italy). The use of animals for these experiments and for those described below was authorized by the ethical committee for animal experiments of the University of Ferrara and by the Italian Ministry of Health (authorization number 36/2008). Hippocampi were dissected and minced with forceps, and then completely dissociated into a single-cell suspension using trypsin digestion. Isolated hippocampal cells were plated at a low density of approximately 5 × 104 cm−2 viable cells in 24-well plates coated with poly-L-lysine (Sigma, St. Louis, MO, USA). Cells were grown in DMEM supplemented with 10% FBS, 50 U/ml penicillin, and 50 mg/ml streptomycin. The culture medium was replaced with the different conditioned media (see below) supplemented with 5 μM arabinofuranosyl cytidine (Ara-C) at the day after plating.
Organotypic Hippocampal Slice Cultures
Hippocampal organotypic slice cultures (OCs) were prepared as described by Stoppini et al. (57) with slight modifications. Male Swiss mice (4 weeks old, Morini Co., Italy) were briefly anesthetized by diethyl ether and decapitated. The brains were removed into ice-cold artificial cerebrospinal fluid (aCSF) consisting of (in mM): NaCl 118, KCl 2.5, MgSO4 3, NaH2PO4 1.1, NaHCO3 26, CaCl2 1, and glucose 11 (all reagents from Sigma) bubbled with 95% O2/5% CO2. Subsequently, 300-μm-thick coronal slices were cut with a vibrotome (MA752, Campden Instruments Ltd., UK). The hippocampi were dissected in cold, oxygenated Hank's balanced salt solution (HBSS, Gibco, Invitrogen), transferred onto sterile porous membrane confetti (Millicell, Millipore, MA, USA), and cultured with their standard medium (here referred to as Medium-2) or with the MAB-conditioned media (see below). The culture medium was changed the day after preparation and then every 2 days for the course of the experiment.
Preparation of the Conditioned Media
The effects of the MABs delivering BDNF on cell survival were evaluated in several experimental groups cultured with a special conditioned media. In the groups “MABs” and “MABs-BDNF,” the conditioned media were composed by the mixture of equal volumes of fresh MABs growing medium (Medium-1) and of the 2-day-culture supernatant from MABs and MABs-BDNF, respectively. The group called Medium-1 served as a control, being challenged with the fresh growing medium. Medium-2 is a standard culture medium for OCs from postnatal animals, consisting of 50% DMEM, 25% horse serum, 18% HBSS, 4 mM L-glutamine, 12 mM glucose, 4.5 mM NaHCO3, 20 mM sucrose, 100 U/ml penicillin, and 100 mg/ml streptomycin (Sigma, St. Louis, MO, USA). The other conditioned media were based on the above media with different supplementations of reagents, like the recombinant human BDNF (0.03–300 ng/ml, Immunological Sciences), the BDNF antagonist K252a (50 nM, Sigma), or TrkB-IgG (2 μg/ml, R&D Systems, Inc., MN, USA), a recombinant tyrosine kinase receptor B (TrkB) engineered as an immunoadhesion to sequester BDNF. Sister slices were randomly assigned to the different groups.
Viability
The viability of PCs and OCs was assessed in two ways. First, the fluorescein diacetate (FDA) hydrolysis assay was used to measure enzyme activity in cells. Slices have been incubated with 10 μg/ml FDA (Sigma) for 30 min, then images were captured using an optical microscope (DMRA2, Leica, Germany), and the fluorescence intensity was quantified using the software Image-Pro Plus 6.0 (Media Cybernetics, USA). Second, the lactate dehydrogenase (LDH) release assay was used to measure cell death. The culture medium was all collected, and LDH leakage was quantified by using a LDH cytotoxic test kit (Clontech, CA, USA) according to the manufacturer's instructions.
Immunocytochemistry
Cultured slices were fixed with 4% PFA in PBS for 1 h at room temperature (RT). After a 48-h incubation with 30% sucrose at 4°C, slices were sectioned to a thickness of 30 μm in a cryostat (CM1510, Leica, Germany). The primary antibody was mouse anti-micro-tubule-associated protein (MAP2abc; 1:50; Immunological Sciences, Italy), and the secondary antibody was Alexa Fluor 488 goat anti-mouse IgG (1:100, Invitrogen, CA, USA). The slices were mounted onto slides after staining with 4′,6-diamidino-2-phenylindole (DAPI, 1:1,000). For zif7268, hippocampal neurons were incubated for 3 h at RT with rabbit anti-zif antibody (Egr-1; Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:1,000 in 10% fetal calf serum (FCS) in PBS, followed by fluorescein anti-rabbit IgG antibody (Invitrogen, CA, USA).
Western Blot
To estimate the density of the surviving neurons within a slice, we performed immunoblot analysis of the neuron-specific marker neurofilament 68. After 14 days in culture, slices were rinsed with ice-cold PBS and then lysed in sample buffer (ECL Western blotting kit, GE Healthcare, USA). Aliquots from each sample (15 μg protein/lane) were subjected to 10% SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidine fluoride membranes (Millipore). The blots were blocked in blocking buffer (20 mM Tris–HCl, 137 mM NaCl, and 5% skim milk) for 1 h at room temperature and then treated with anti-neurofilament 68 antibody (diluted 1:500; Sigma) overnight at 4°C. Membranes were washed repeatedly in Tris-buffered saline containing 0.05% Tween 20, and then the horse radish peroxidase-conjugated secondary antibody (diluted 1:20,000) was added for 1 h. Immunoreactive bands were detected using enhanced luminol-based chemiluminescence (ECL). The membranes were stripped, and then immunoblotting for β-actin (1:1,000, Sigma, St. Louis, MO, USA) was performed, as a loading control. Bands were scanned into digital images and analyzed with the software of Image-Pro Plus (Media Cybernetics, USA).
Field Potential Recording
After 7 days in culture, slices were transferred to a holding chamber for 1 h at room temperature in aCSF, while continuously aerated with 95% O2 and 5% CO2. Slices were then placed in a submerged in vitro recording chamber and perfused with oxygenated aCSF. The temperature in the recording chamber was kept at 36 ± 1°C. Bipolar wire electrodes (tungsten with a tip diameter of 90 μm; WPI, Inc., FL, USA) were used for stimulation of the Schaffer collateral pathway in the CA3 region. Glass microelectrodes filled with 0.9% NaCl were placed on the CA1 stratum radiatum to record field excitatory postsynaptic potential (fEPSP). A conventional electrophysiological technique for extracellular recordings was employed to identify the maximal response and to adjust the stimulus strength. The stimulus intensity (70-μs duration rectangular pulses at 40 V) that repeatedly evoked the maximal synaptically evoked response/excitatory postsynaptic potential was used in each slice. Signals were acquired under constant conditions and off-line processed using the Patchmaster software (HEKA Instruments, Inc., Germany).
Statistical Analysis of Data
One-way analyses of variance (ANOVA) followed by the Newman–Keuls test were used to evaluate the results. Effects were considered to be statistically significant at the 0.05 probability level.
Results
Selection and Characterization of Mesoangioblast Cells Expressing NGF and BDNF
Two DNA constructs were generated, driving the expression of murine preproNGF and rat preproBDNF. The preproNGF construct contains the cDNA encoding the short form (27 kDa) of the NGF precursor (isolated from the mouse submaxillary gland), whereas the preproBDNF construct contains the cDNA coding for the BDNF precursor (isolated from the rat brain). These constructs are based on the scFv-express vector (50), yielding the expression vectors ScFv-preproNGF and ScFv-preproBDNF, respectively. In order to evaluate the appropriate expression and targeting to secretion of the pro-neurotrophins driven by the constructs, COS cells were transiently transfected with ScFv-preproNGF or ScFv-preproBDNF. NGF and BDNF expression and secretion in the COS medium were assessed 72 h after transfection using ELISA. The average concentration of NGF was estimated to be approximately 80 ng/ml and that of BDNF was approximately 190 ng/ml (data not shown).
GFP-expressing MABs (45) were stably transfected with either ScFv-preproNGF or ScFv-preproBDNF DNA. Clonal selection was performed with G418 and limited dilution. Among the different clones analyzed by ELISA for neurotrophin production, two were selected: clone F10, producing the highest concentration of NGF after 4 days in culture, and clone A9, producing the highest concentration of BDNF after 15 days in culture. These clones were then further characterized for the kinetics of NGF and BDNF secretion. For this purpose, conditioned medium was collected from F10 cultures kept at confluence (3.5 × 104/cm2) 24, 48, 72, and 96 h (4 days) in the case of NGF; in the case of BDNF-expressing MABs (clone A9), mitomycin-treated cells were kept for 8, 11, 14, and 15 days. Within these time frames, the maximal concentration of NGF was reached after 4 days of culture and the one of BDNF after 15 days. BDNF secretion was very low in the first week. The reason(s) for the different kinetics of NGF and BDNF secretion are presently under investigation. The estimated rate of accumulation was 36 ng/ml/day/106 cells for NGF and 30 ng/ml/day/106 cells for BDNF, starting from the 8th day in culture.
To determine if the proneurotrophins were successfully processed by intracellular convertases and if the secreted forms are indeed mature NGF or BDNF, conditioned media collected from control and transfected cells were analyzed. Media from F10 MABs-NGF and A9 MABs-BDNF, as well as from control nontransfected MABs, were analyzed by Western blot. Only mature NGF was detected in the medium of F10 MABs-NGF (Fig. 1A). In the medium of A9 MABs-BDNF, proBDNF was detectable but in much smaller amounts compared with mature BDNF (Fig. 1B). These data indicate that MABs-NGF and MABs-BDNF can process the proneurotrophins and secrete exclusively or almost exclusively the mature form.
Assessment of NGF Biological Activity
The biological activity of NGF was evaluated using the PC12 cell differentiation bioassay (19). Duplicates of PC12 cells were challenged with the conditioned medium from NGF-producing MABs (clone F10) kept in culture for 5 days (containing approximately 80 ng/ml NGF, based on ELISA), diluted 1:1 with growing medium. In alternative, PC12 cells were cocultured with F10 cells in a 1:1 ratio. The medium was replaced every other day, and PC12 cell differentiation was assessed by the presence of neurite elongations and the expression of the neuronal marker β-tubulin after 7 days. As shown in Figure 2, both F10 conditioned medium and coculture with F10 cells induced efficient neuronal differentiation of PC12 cells similar to that observed upon the addition of recombinant NGF (50 ng/ml). Neither the conditioned medium from control, untransfected MABs, nor coculture with these cells induced PC12 differentiation.
Production and release of mature neurotrophins by mesoangioblasts (MABs). (A) Western blot analysis for nerve growth factor (NGF) shows the mature form of the neurotrophin (left arrow) in the supernatant of NGF-expressing F10 cells (MABs-NGF) but not in normal MABs. The amount of proNGF in the medium is negligible. (B) Western blot analysis for brain-derived neurotrophic factor (BDNF) shows the mature form of the neurotrophin (left arrow) in the supernatant of BDNF-expressing A9 cells (MABs-BDNF) but not in normal MABs. proBDNF is detectable in the medium, but in much smaller amounts compared with BDNF, indicating a nearly complete processing. Multiple bands can be observed for BDNF, likely corresponding to posttranslational processing products. Effects of NGF produced by MABs on PC12 differentiation. PC12 differentiation in the presence of recombinant NGF (B, E, H, K), in the presence of conditioned medium from MABs-NGF (C, I), or when cocultured with MABs-NGF (F, L) is visualized by the presence of neurites (B–C, E–F) and confirmed by immunofluorescence for the expression of β III-tubulin (Tuj1, red immunostaining in H–I, K–L). As negative control, PC12 cells have been cultivated in growing medium without NGF (A, G) or cocultured with control MABs (D, J). Scale bars: 25 μm.

Assessment of BDNF Biological Activity
The biological activity of BDNF was evaluated in several ways. First (Fig. 3), we measured the nuclear accumulation of zif/268 in hippocampal primary cultures (PCs). Zif/268 (Egr1) is a zinc finger protein acting as transcription factor, whose nuclear translocation is commonly used to test the biological activity of BDNF (51). PCs were challenged for 3 h with the conditioned medium from MABs-BDNF (A9 clone), collected after 15 days of culture and containing approximately 87 ng/ml BDNF. Immunofluorescence revealed that the nuclear accumulation of zif/268 was increased upon addition of A9 conditioned medium (Fig. 3D), indistinguishable from what was observed upon addition of 50 ng/ml recombinant BDNF (rBDNF) (Fig. 3B), as compared with untreated neurons (Fig. 3A) and neurons treated with medium conditioned from control MABs (Fig. 3C).
Effects of BDNF on zif/268 nuclear accumulation in primary rat hippocampal neurons. High levels of zif/268 immunostaining are concentrated in the nuclei of cells incubated with recombinant BDNF (arrows in B) or conditioned medium from A9 BDNF-producing MABs (arrows in D), whereas low level of zif/268 immunostaining is present in the nuclei of hippocampal neurons treated with growing medium (A) or medium conditioned from control, D16 MABs (C). Scale bars: 25 μm.
Second, we tested if continuous application of rBDNF in the medium exerted trophic effects on cell survival in low-density PCs and in organotypic cultures of adult hippocampus slices (OCs). Thus, culture media were supplemented with increasing concentrations of rBDNF, ranging from 0.03 to 300 ng/ml. In PCs, neurons seeded at low density can hardly survive without the support of rBDNF. Quantification of survival, based on cell counting and on LDH release, showed that BDNF produces a significant beneficial effect in a concentration-dependent manner (Fig. 4A, B). The maximal effect was estimated to be reached at a concentration of approximately 10−8 M (corresponding to 270 ng/ml) and the EC50 was approximately 3 × 1 0−11 M (slightly less than 1 ng/ml). These data are in keeping with previous findings (6,37).
Recombinant BDNF enhances cell survival in primary hippocampal neuronal cultures (PCs, A and B) and in organotypic adult hippocampus slice cultures (OCs, C and D) in a concentration-dependant manner. Neurons were cultured with the indicated concentration of BDNF for 7 days, slices for 14 days. A and C indicate viable cells, as quantified using cell counting for PCs and fluorescence intensity of the marker fluorescein diacetate (FDA) for OCs. B and D represent cell death, as estimated measuring the levels of released lactate dehydrogenase (LDH). Data are expressed as the means ± SE (n = 6). *p < 0.05, **p < 0.01 versus control (0 ng/ml); ANOVA and post hoc Newman–Keuls test.
We then examined the neurotrophic activities of rBDNF in OCs. OCs provide an ideal in vitro model system to assess toxic or trophic effects since they preserve the morphological and physiological features of the hippocampal neuronal network and allow easy access and precise control of extracellular environment for a long period (28,57). In general, OCs are prepared from early postnatal animals (14) because, when prepared from adult hippocampal slices, they rapidly undergo neuronal degeneration (64). We elected to employ hippocampal cultures prepared from adult animals (66) exactly for this reason: they do not survive well in standard culture media and, thus, may provide a very convincing evidence for the favorable effects of the supplementation of NTFs if they survive when cultured in the MAB medium (in this sense, OCs represent an in vitro model of neurodegeneration).
In the absence of rBDNF, OCs displayed degeneration aspects (like white spotted cell debris and uneven surface) after 6–8 days in vitro (DIV), which were clearly visible under phase contrast microscopy. Similar to PCs, rBDNF concentration-dependently reduced these signs of degeneration. Viability was quantified using FDA fluorescence to identify viable cells and LDH release to estimate the degree of cell death. As shown in Figure 4C and D, BDNF concentration-dependently increased FDA fluorescence and reduced LDH release, in line with previous observations reporting that BDNF enhances the cell tolerance to serum deprivation of organotypic cultures from postnatal slices (48). These effects were obtained in the same range of concentrations that proved effective for PCs.
As stated above, the amount of BDNF secreted into the medium (Medium-1) from MABs-BDNF after 2 days in culture was very low: approximately 2 ng/ml, as measured using ELISA (Fig. 5). If MABs-BDNF were cultured with the medium usually employed to culture OCs (Medium-2), the amount of secreted BDNF was approximately halved (Fig. 5C). Therefore, although controls were also performed using Medium-2, the conditioned media that we used in all experiments was based on Medium-1: 50% culture supernatant from 2-day-cultured MABs-BDNF and 50% fresh Medium-1. This conditioned medium contained approximately 1 ng/ml BDNF, whereas no detectable of BDNF was present in the medium conditioned with control MABs (Fig. 5C).
MABs-BDNF deliver BDNF and enhance cell survival in primary hippocampal neuronal cultures (PC) and in organotypic adult hippocampus slice cultures (OC). (A and B) Genetically modified MABs-BDNF proliferate and express green fluorescent protein (GFP). (C) MABs-BDNF secrete BDNF. Concentration of BDNF in the medium of MABs and MABs-BDNF at various days in vitro (DIV), as measured using ELISA. MABs-BDNF were cultured in two different types of medium (Medium-1 and Medium-2, see text for details) as indicated. (D) Effect of different treatment procedures on the viability of hippocampal PCs, as estimated using the LDH release assay. Data are the means ± SE of six separate experiments. **p < 0.01 versus Control-1; •p < 0.05 versus MABs; ANOVA and post hoc Newman–Keuls test. (E) Effect of different treatment procedures on the viability of adult hippocampus OCs, as estimated using the LDH release assay. Data are the means ± SE of six separate experiments. *p < 0.05 and **p < 0.01 versus Control-1; •p < 0.05 versus MABs; ▴p < 0.05 versus MABs-BDNF; ANOVA and post hoc Newman–Keuls test. Control-1 are slices treated with the typical medium for MABs culture (Medium-1). Control-2 are slices treated with the typical medium for postnatal slice culture (Medium-2). Scale bars: 25 μm (A and B).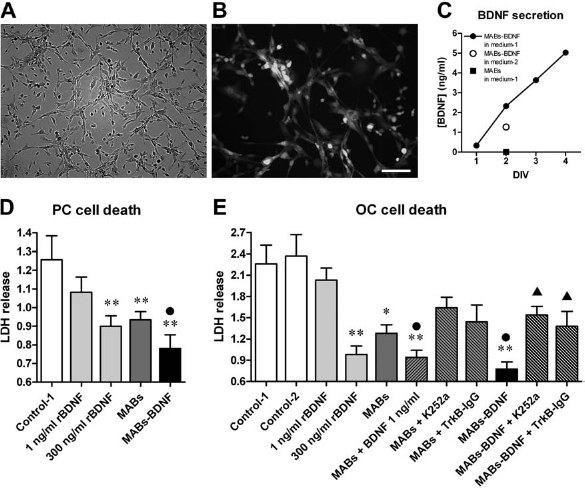
We first screened the trophic effects of the MABs-BDNF-conditioned medium in the PC system. The medium conditioned in control MABs increased the number of surviving neurons by about 80%, similar to the effect of 300 ng/ml rBDNF (a maximally effective concentration). The effect of the MABs-BDNF-conditioned medium was much greater (it enhanced neuronal survival by approximately 170%). We used the LDH assay to measure these effects: as shown in Figure 5D, both 300 ng/ml rBDNF and the MABs medium significantly (and to a similar extent) reduced cell death, but the MABs-BDNF medium had a significantly greater effect. Interestingly, 1 ng/ml rBDNF (i.e., the concentration of BDNF present in the MABs-BDNF medium) alone did not produce any significant effect on neuronal survival. Together with the observation of a significant effect of the MABs medium, this indicates that other trophic factors are secreted by MABs that can synergize with BDNF to produce neuroprotection.
We then extended and deepened this analysis in OCs. Adult OCs were cultured with the different media, and after 14 DIV, cell death was measured using the LDH release assay. As shown in Figure 5E, cell death was remarkable in control media (Medium-1 and Medium-2). As with PCs, the MABs-conditioned medium significantly decreased cell death to nearly the level of rBDNF 300 ng/ml, and the MABs-BDNF-conditioned medium produced a significantly more pronounced effect. This extra effect is mediated by the low concentrations (1 ng/ml) of mature BDNF produced by the MABs-BDNF, because (i) adding 1 ng/ml rBDNF to the MABs medium produced an effect similar to the one produced by the MABs-BDNF medium; (ii) adding the TrkB inhibitor K252a and the BDNF scavenger TrkB-IgG to the medium conditioned by MABs-BDNF abolished the extra effect observed in the MABs-BDNF group, while these two BDNF blockers had no significant influence on the medium conditioned by MABs (Fig. 5E). These data support the notion that the prosurvival effect of the MABs-BDNF medium is mediated by a synergy between BDNF and other, yet unidentified, soluble substances constitutively produced by MABs. Accordingly, 1 ng/ml rBDNF alone did not reduce cell death at all, and much higher concentrations (300 ng/ml) were needed to approach the effect of the MABs-BDNF-conditioned medium.
These findings have been confirmed using light microscopy and FDA fluorescence, an indicator of viable cells (Fig. 6). At the bright field observation, as compared with slices cultured in the control medium (these slices, as described, display obvious signs of degeneration) (Fig. 6A), the slices cultured in the medium conditioned on MABs-BDNF remained thicker and preserved their structural organization for a long period, up to 25 days (Fig. 6C). Slices cultured in the MABs medium appeared healthier than those cultured in control medium but less healthy than those cultured in the MABs-BDNF medium (Fig. 6B). In line with these findings and paralleling the high levels of LDH release described above, FDA fluorescence was weak and uneven in OCs cultured with control medium (Fig. 6D), indicating that most of the cells are indeed degenerating. OCs cultured with the MABs medium displayed a better labeling quality (Fig. 6E), which was further improved in OCs cultured with the MABs-BDNF medium (Fig. 6F).
Morphological evidence of the beneficial effects of the medium conditioned in MABs-BDNF on adult organotypic cultures. Adult slices were cultured for 14 days with the control Medium-1, with medium conditioned in MABs, or with medium conditioned in MABs-BDNF. Note that slices cultured in the MABs-BDNF-conditioned medium, compared with the other two groups, better maintain their integrity based on bright field observation (A–C) and remain more viable based on FDA (D–E), DAPI staining (G–I), and microtubule-associated protein 2 (MAP2) immunohistochemistry (J–L). Nuclei in blue in G–I; neurons in green in J–L. Scale bar: 1 mm (scale bar in C also refers to A and B; scale bar in F also refers to D and E; scale bar in I also refers to G and H; scale bar in L also refers to J and K).
Cell counting, performed by using the DAPI nuclear staining, confirmed that, even if some cell loss was present in slices cultured with MABs-BDNF-conditioned medium (Fig. 6I), the number of surviving cells was much higher than in the control group (Fig. 6G). Slices treated with the medium conditioned in MABs performed a little better than controls, but much worse than those treated with the medium conditioned in MABs-BDNF (Fig. 6H).
Other morphological examinations and electrophysiological recordings were performed. For morphological analysis, slices were fixed and immunostained with MAP2, a cytoskeletal protein primarily found in neuronal dendrites. As shown in Figure 6J, adult slices cultured in the control media for 14 days exhibit not only extensive cell loss, but also a grossly altered structure, with many lacunae in the pyramidal cell layer and just a few remaining, exclusively pyknotic neurons. MAP2 staining was essentially absent in control slices. While the MABs medium only slightly improved this situation (Fig. 6K), the MABs-BDNF-conditioned one clearly attenuated neuronal loss (Fig. 6L). Furthermore, the dendritic network and the cytoarchitectonic characteristics were essentially maintained in adult OCs treated with the MABs-BDNF-conditioned medium, even if some signs of neuronal degeneration could be detected (loss of neurons, decreased dendrites, condensed or swelling pyramidal cells). These signs of degeneration progressively increased in time. This morphological analysis further confirms that the medium conditioned in MABs-BDNF enhances neuronal survival and delays the alterations in structural organization.
The density of neurons in OCs was also assessed using Western blot analysis for the neuron-specific marker neurofilament 68 (NF68). We found that OCs cultured on MABs-BDNF-conditioned medium contained more NF68, thus more neurons, as compared with slices cultured in the control medium or in medium from conventional MABs (Fig. 7). A statistically significant difference was also found between MABs and control group, indicating that supplementation of the soluble substances produced by MABs can slightly attenuate neuronal loss.
Density of neurons surviving in adult slices after 14 days in vitro under the different experimental conditions. Neuronal density was quantified performing Western blot analysis for a neuron-specific marker, neurofilament 68. (A) Representative blot. (B) Data quantification. The data are means ± SE of four separate experiments. *p<0.05 and **p<0.01 versus Control-1; ••p<0.01 versus MABs; ANOVA and post hoc Newman–Keuls test.
Finally, we performed electrophysiological recordings to demonstrate the viability of the surviving neurons and the persistence of synaptic connections at a functional level. Thus, fEPSPs in CA1 pyramidal neurons have been measured after stimulation of the Schaffer collaterals. We chose 7 DIV as a check time point, because adult slices in culture, even those cultured with the MABs-BDNF-conditioned medium, display a progressive decrease in the amplitude and stability of the extracellular field potentials in time (data not shown). Such currents could not be reliably measured at 14 DIV, when neurons are still visible using immunofluorescence: as expected, the functional damage precedes the morphological one. As shown in Figure 8A, adult slices cultured with the typical medium exhibited very small (if any) synaptic response at 7 DIV, while relatively stable fEPSPs could be recorded in those cultured with the medium conditioned in MABs or MABs-BDNF. However, the mean amplitude of the evoked fEPSPs in the MABs-BDNF group was much higher than in the MABs group (Fig. 8B). The presence of evoked synaptic responses, albeit lower and less stable than in acute slices or in cultured postnatal slices, demonstrates that the neuronal activity and the hippocampal circuitry remain functional by virtue of the beneficial effects of the mediators secreted by MABs-BDNF.
Maintenance of synaptic connections in adult slices after 7 days in culture with the MABs-BDNF-conditioned medium. Synaptic connections were identified by recording evoked field excitatory postsynaptic potential (fEPSP) in CA1 after stimulation of the Schaffer collaterals. (A) Representative recordings of the evoked fEPSP in the different groups. (B) Peak amplitude of the fEPSPs, measured and statistically analyzed as described in Materials and Methods. The data are the means ± SE of five separate experiments. **p<0.01 versus Control-1; ••p<0.01 versus MABs; ANOVA and post hoc Newman–Keuls test.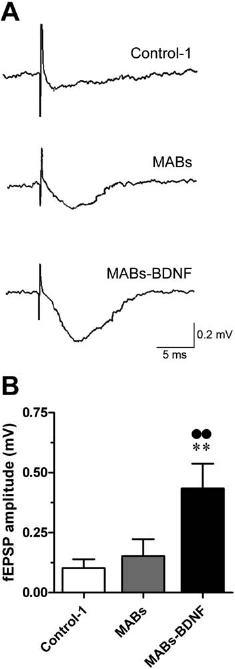
Discussion
Main Findings
In this study, we generated MABs that can stably secrete the neurotrophins NGF or BDNF in their mature form and found that media conditioned from the culture supernatant of these cells exert trophic effects in vitro: PC12 cells are induced to differentiation by a MABs-NGF-conditioned medium, while viability and function of cultured neurons and brain slices are increased by a MABs-BDNF-conditioned medium. MABs-BDNF effects are TrkB receptor-dependent, being abolished by the receptor blocker K252a or by the BDNF scavenger TrkB-IgG. Thus, MABs produce robust bystander effects on the nervous tissue that we characterized using endpoints (neuronal differentiation, neuroprotection, function) that are relevant for neurodegenerative diseases (or, more in general, neurological diseases associated with damage).
Therapeutic Efficacy of NGF and BDNF in Neurodegeneration
A large body of evidence shows the essential role played by NGFs in the adult CNS. In particular, basal forebrain cholinergic neurons (BFCNs) that are selectively vulnerable and hit in AD (1) depend on NGF for their function and maintenance of cholinergic phenotype (17,55). BFCNs express NGF receptors (21,22), retrogradely transport NGF from their cortical projections up to their cell bodies (12), and respond to administration of exogenous NGF increasing cholinergic markers (17,46). Moreover, NGF can prevent BFCNs death or atrophy (21,29,63,65).
A pivotal protective role of NGF in neurodegeneration has also been shown in AD patients. Clinical trials are currently underway using intracerebral delivery systems that by-pass the BBB. For example, a phase 1 clinical trial used stereotaxic intracerebral transplants of autologous fibroblasts engineered to produce NGF, achieving a consistent reduction in the progression of cognitive decline (62). Promising results from another Phase 1 clinical trial, evaluating the safety and efficacy of stereotactic intracerebral injections of adeno-associated virus (AAV) harboring the NGF gene have recently been reported (41). Unfortunately, an invasive surgical step is required for these approaches. One alternative may be the delivery of NGF through the olfactory route, a less invasive approach that proved capable to achieve pharmacologically relevant NGF concentrations in the brain of animals (3,9,10). In one study (10), nasal NGF delivery reverted the neurodegenerative phenotype in an Alzheimer's disease (AD) model, in the absence of appreciable side effects. The drawback of this approach is that it requires repeated injections for a prolonged period.
BDNF is also known to exert neuroprotective effects. For example, exogenous BDNF has been reported to increase survival of neurons in cultures, while reduced survival was found after addition of anti-BDNF antibodies and in BDNF knockout mice (38,43). BDNF appears to be an essential constituent of the microenvironment for neuronal survival in the adult hippocampus. It potentiates presynaptic release and excitatory transmission (24,25,35) and promotes dendritic and axonal growth (30). Moreover, several investigations indicate a pivotal role of BDNF in neuroprotection in different in vivo models of disease, including ischemia (27,32) and seizures (18,31). The signaling mechanisms underlying the prosurvival effects of BDNF appear to involve TrkB activation. TrkB receptors have long been known to mediate trophic support of adult neurons (13), and a number of studies show that decreased expression of the receptor is associated with cell loss (44). Coherently with these data, we demonstrated here the beneficial effects of rBDNF or MABs-delivered BDNF on cell survival in primary neuronal and in adult slice cultures, effects that were TrkB receptor-dependent. We also observed that MABs-delivered BDNF facilitates synaptic responses, an effect that may be ascribed to a better preservation of synaptic connection or to a potentiation of synaptic transmission. It has been reported that BDNF modulates the strength of existing synaptic connections (24,25) and favors the formation of new synaptic contacts within the hippocampal circuits (33).
Similar to NGF, clinical trials have been also undertaken with BDNF (both peripherally administered and delivered directly in the CNS) for the treatment of neurological diseases (56,60). Unfortunately, data have been negative thus far.
Synergy Between MAB-Produced Trophic Factors
We found that the amount of BDNF released by MABs-BDNF produced greater effects than the identical amount of rBDNF, suggesting either that the specific activity of MAB-released BDNF is higher than that of rBDNF (possibly due to a better folding of BDNF released by MABs) or that other neurotrophic factors produced by MABs synergize with BDNF. A microarray analysis of gene expression in MABs revealed that several growth factors are expressed at high level in these cells, including VEGF B, FGF-2, FGF-7, PDGF AA, and others (15). Although it remains unknown which of these NTFs may exert neuroprotective effects, it seems very likely that they will interact and possibly synergize with BDNF.
Regardless of the underlying mechanisms, the markedly beneficial effects of MABs-delivered NTFs on neurons and brain tissue suggest that exogenous, MAB-mediated, administration of BDNF or NGF (maybe synergizing with other MAB-produced NTFs) might be developed into a strategy for functional preservation and restoration in neurological diseases associated with brain damage. In this prospect, a very attractive property of MABs is their potential to reach the lesioned tissue when peripherally administered, because this would represent a net advantage over other currently tested delivery strategies. MABs have been reported to home in damage areas like skeletal muscle (53) and myocardial tissue (15) after systemic infusion.
Conclusions: Potential Advantages of MABs as a Delivery Strategy
The supplementation of NTFs can sustain the survival and functional recovery of neurons by modulating the postinjury microenvironment. However, NTF delivery to the nervous system should be target-specific and regionally restricted, prolonged (although not necessarily lasting for life), safe, well tolerated, and sufficiently robust to elicit responses from target neurons. MAB-based NTF delivery may be optimal to meet these criteria. MABs possess the advantages of all cell-based therapies: they can be expanded to virtually unlimited numbers in vitro; they are amenable to genetic modification; they have self-renewal and multipotent capacities. Moreover, (1) they may be used for autologous transplants (can be prepared from biopsies from the patient himself); (2) they may be peripherally administered (no need for surgery); (3) they may selectively home in the lesioned area (recruitment at the neuroinflammated site), with no undesired accumulation of NTF in areas that do not need them (reduced side effects); (4) at the site of accumulation, they may act as efficient bioreactors producing robust bystander effects.
Further studies will be required to verify the selective homing of MABs in lesioned areas after peripheral administration. If these new studies will be successful, MABs might be developed into a new cell-based strategy for NTF delivery into the CNS, promoting neuronal survival and restoring network function in neurological diseases associated with brain damage.
Footnotes
Acknowledgments
The authors thank J. Tonnesen and M. Kokaia for the useful advices on OCs; M. Urso and L. Pistillo for technical help; P. Marconi for Western blot analysis on BDNF; M. Righi for rat hippocampal primary cultures preparation and anti-zif immunofluorescence; S. Capsoni for assistance and advise on the use of AD11 mice and for suggestions concerning the in vivo olfactory delivery; F. Paoletti and M. Benedetti for providing preproNGF and preproBDNF cDNAs, respectively; E. Margotti and S. Covaceuszach for preparing TrkB immunoadhesin; and M. Gulisano for scientific discussions. This work was supported by grants from the University of Ferrara (FAR, to MS), the Italian Ministry for the University and Scientific Research (Prin 2007 to MS and AC; FIRB RBIN04H5AS to AC), the European Community [LSH-CT-2006-037315 (EPICURE), thematic priority LIFESCIHEALTH, to MS], and the Compagnia di San Paolo (to MS). LF was partly funded by SISSA/ISAS, Trieste. The authors declare no conflicts of interest.