
Abstract
Among the animal models of Duchenne muscular dystrophy (DMD), the Golden Retriever muscular dystrophy (GRMD) dog is considered the best model in terms of size and pathological onset of the disease. As in human patients presenting with DMD or Becker muscular dystrophies (BMD), the GRMD is related to a spontaneous X-linked mutation of dystrophin and is characterized by myocardial lesions. In this respect, GRMD is a useful model to explore cardiac pathogenesis and for the development of therapeutic protocols. To investigate whether cardiac progenitor cells (CPCs) isolated from healthy and GRMD dogs may differentiate into myocardial cell types and to test the feasibility of cell therapy for cardiomyopathies in a preclinical model of DMD, CPCs were isolated from cardiac biopsies of healthy and GRMD dogs. Gene profile analysis revealed an active cardiac transcription network in both healthy and GRMD CPCs. However, GRMD CPCs showed impaired self- renewal and cardiac differentiation. Population doubling and telomerase analyses highlighted earlier senescence and proliferation impairment in progenitors isolated from GRMD cardiac biopsies. Immunofluorescence analysis revealed that only wt CPCs showed efficient although not terminal cardiac differentiation, consistent with the upregulation of cardiac-specific proteins and microRNAs. Thus, the pathological condition adversely influences the cardiomyogenic differentiation potential of cardiac progenitors. Using PiggyBac transposon technology we marked CPCs for nuclear dsRed expression, providing a stable nonviral gene marking method for in vivo tracing of CPCs. Xenotransplantation experiments in neonatal immunodeficient mice revealed a valuable contribution of CPCs to cardiomyogenesis with homing differences between wt and dystrophic progenitors. These results suggest that cardiac degeneration in dystrophinopathies may account for the progressive exhaustion of local cardiac progenitors and shed light on cardiac stemness in physiological and pathological conditions. Furthermore, we provide essential information that canine CPCs may be used to alleviate cardiac involvement in a large preclinical model of DMD.
Introduction
Dystrophinopathies are a group of inherited diseases characterized by the absence of structural proteins of the dystrophin glycoprotein complex (DGC). Duchenne (DMD) and Becker muscular dystrophies (BMD) are caused by mutations in the dystrophin gene, which encodes for the largest protein in the DGC and primarily effects skeletal and cardiac muscle (13). Both DMD and BMD are associated with cardiac degeneration and morbidity leading to hypertrophic then/or dilated cardiomyopathy (2,36). The improvement of multidisciplinary patient care has increased life expectancy in DMD and BMD patients; however, heart failure is now the main cause of death (17,40,52). Thus, rescuing dystrophin expression in cardiac muscle has become more critical for the longevity and quality of life in DMD and BMD patients. Among the various animal models of DMD, the Golden Retriever muscular dystrophy (GRMD) dog is considered the best model in terms of size and pathology onset. The GRMD dog carries a point mutation in the splice acceptor site in intron 6 of the orthologous X-linked dystrophin gene (18) and has a clinical course very similar to that of human patients, characterized by progressive muscle wasting, degeneration, fibrosis, and a shortened lifespan (3,22,27). Cardiac involvement in GRMD dogs has been demonstrated by electrocardiographic studies, with the onset of a progressive cardiomyopathy similar to DMD patients (10,38,54). In this respect, GRMD is a useful model to explore cardiac pathogenesis and for the development of new therapeutic protocols (56). Although considered the closest animal model to humans, the canine model has been poorly described in respect of stem cell therapy for chronic and acute cardiac diseases. More importantly, a recent study in dystrophic mice suggests that dystrophin restoration may exacerbate the failure of heart function if protein expression is not effectively restored in cardiac muscle (53). Recently, it has been demonstrated that the presence of cardiac stem cells, including mesoangioblasts, from mice and human cardiac biopsies are able to differentiate spontaneously in vitro and in vivo toward the cardiomyogenic lineages (19,20,29,31,37,39,41). To date, no comparative study has been performed to test the regenerative potential of these progenitors isolated from the dystrophic mice model. Here we describe the isolation of self-renewing cardiac progenitors (CPCs) from different regions of wild type and GRMD heart, capable of contributing to cardiovascular lineages in the postnatal forming heart. Under particular stimulation CPCs form smooth muscle, endothelial cells and immature cardiomyocytes, albeit with varying capacity. Interestingly, GRMD cardiac progenitors showed limited lifespan and proliferation deficiency compared to the wt counterpart.
Materials and Methods
Cell Culture
CPCs were isolated from cardiac muscle biopsies of GRMD and wild-type golden retrievers. GRMD and healthy dog colonies were maintained at the Veterinary School of Maison-Alfort (Paris, France; see In Vivo Experiment section). Biopsies from different heart regions were kept in DMEM 10% fetal bovine serum (FBS) and separated by their regions of origin, namely aorta, atrium, and ventricle. Most fragments contained small vessels (selected under microscope) and were transferred to a Petri dish coated with 1% gelatin or collagen in presence of 5% FBS MegaCell medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 5 ng/μl basic fibroblast growth factor (bFGF) plus 5 mM l-glutamine and antibiotics (Invitrogen, Carlsbad, CA, USA). These heart fragments were cultured for 8–12 days depending on the region and after the initial outgrowth of fibroblast-like cells, small round and refractile cells appeared. This cell population was easily collected by gently pipetting off the original culture, then counted and cultured in T75 flasks (Nunc, Rochester, NY, USA) using MegaCell medium. Single clones from each cardiac biopsy were obtained by serial dilution of three cells per 96 wells. Firstly, growing clones were selected morphologically and further analyzed as discussed in the Results section. Four clones from each cardiac biopsy (atrium, aorta, and ventricle) were taken into account for the analysis. For cell culturing, 103 cells/ cm2 were plated in growing medium and incubated until they reach 75% confluency (usually 4 × 104 cells/cm2). Population doubling (PD) time was calculated as the ratio Δt*(log2/logNt - logN0), where Δt = period time, N = number of cells at time t, and N0 = number of cells at time 0. For coculture experiments, neonatal rat cardiomyocytes were isolated using the Neonatal CM isolation kit (Worthington Biochemical Corporation, Lakewood, NJ, USA) according to the manufacturer's protocol. Mouse myogenic cell line C2C12 was purchased from ATCC (LGC Standards, Middlesex, UK) and maintained in DMEM supplemented with 5 mM l-glutamine and antibiotics (all from Invitrogen, Carlsbad, CA, USA) and 10% FBS (Lonza). Neonatal rat cardiomyocytes were cultured in DMEM 10% FBS (Invitrogen, Carlsbad, CA, USA). All cultures were incubated at 37°C in a humidified incubator with 5% CO2 and 95% air. Cells were transduced with third generation lentiviral vector expressing green fluorescent protein (GFP) under the control of human phosphoglucokinase (hPGK) promoter as previously described (39). For GFP+ enrichment, ventricle (Ven) CPCs were sorted as described in the next section.
Transfection and Fluorescence Activated Cell Sorting (FACS)
For transfection, 1.5 × 105 canine CPCs were transfected with 1 μg of pCMV-SPORT6/hNkx2.5 (cytomegalovirus vector containing human NK2 homeobox 5) using Effectene reagent (Qiagen, Valencia, CA, USA) according to manufacturer's protocol. For transposon experiments, nucleofection (Lonza, Rockland, ME, USA) was used for transfecting 1 × 106 ventricle CPCs in T75 flasks. Each cell population was transfected with 6 μg of dsRed-containing transposon and 12 μg of transposase vectors derived from the PiggyBac system. For dsRed+ positive enrichment, 107 transposon-transfected Ven CPCs were sorted for dsRed detection by flow cytometry (BD FACSDiva Option).
Immunofluorescence and Western Blot Analysis
For immunocytochemistry, clones were washed with PBS and fixed with 4% paraformaldehyde (PFA) for 10 min. Cells were then stained with antibodies against different molecules overnight at 4°C and detected using species-specific secondary antibodies. Nuclei were counterstained with DAPI (Sigma-Aldrich, St. Louis, MO, US). For tissue histology, mice previously injected with 2.5 × 105 Ven wt or GRMD canine CPCs were sacrificed, and hearts were quickly frozen in OCT (Sakura Finetek Tissue-Tek, Torrance, CA, USA). Serial heart sections were fixed with 4% PFA, permeabilized with 0.1% Triton X-100, rinsed with 1% bovine serum albumin (BSA) in PBS, and immunostained with anti-dsRed, anti-cMyHC (β-cardiac myosin heavy chain) and anti-αSA (sarcomeric α-actinin). Alexa 488 or 594 (Molecular Probes, Eugene, OR, USA) were used as secondary staining Ig, and DAPI was used for nuclear staining. For Western blot analysis, cell pellets were suspended in a lysis buffer (pH 7.5) containing 50 mM Hepes, 150 mM NaCl, 100 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 0.5% Triton X-100, 0.5% Nonidet P-40 (NP-40), 1 mM dithiothreitol, and 10% glycerol and supplemented with a protease inhibitor cocktail (Roche). Whole-cell lysates or tissue extracts were then centrifuged at 17,000 × g for 10 min at 4°C to remove cell debris. Protein extracts (20–30 μg) were electrophoresed on 10% sodium dodecyl sulfate (SDS) polyacrylamide gels under reducing conditions and subsequently transferred onto Immobilon-P membranes (Millipore Chemicon, MA, US) with a semidry blotting apparatus (BioRad, Herts, UK). The membranes were blocked in a 5% skim milk Tris-buffered saline–Tween 20 solution (TBS-T) and subsequently probed with various antibodies. The membrane was washed with TBS-T and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (goat anti-mouse IgG or anti-rabbit IgG antibodies from Santa Cruz) for 30 min. Following further wash steps, the protein bands were visualized by enhanced chemiluminescence (Amersham, GE Healthcare, Diegem, BE) using a Fuji Scanner. Phase contrast images were taken with an inverted fluorescence microscope Nikon ECLIPSE TS-100. Immunofluorescence staining analysis was performed using Nikon ECLIPSE Ti-S inverted microscope. Confocal images were taken with Leica TCS SPE or with an inverted deconvolution microscope (DeltaVision RT) for the paneling images. Image editing was performed using “ImagePro” software provided by Nikon.
Cellular Biochemistry
For alkaline phosphatase (AP) staining, cultured plates or explants were rinsed with PBS fixed in 4% formaldehyde and stained for 1 h in a 0.1 M Tris–HCl solution (pH 8.5) containing 0.2 mg/ml of Napthol AS-MX phosphate and 0.6 mg/ml of Fast Blue BB salt (Sigma-Aldrich, St. Louis, MO, USA). For telomerase analysis, cell pellets at early, mid, and late doublings were harvested, and telomerase activity was measured using TRAPeze RT Telomerase Detection Kit (Millipore Chemicon, MA, USA). Briefly, cell lysates were obtained using CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) lysis buffer and normalized. Following qPCR with specific Amplifluor primers, average Ct values of each sample were plotted to determine the log copy number of telomeres and relative telomerase activity values. For proliferation assays, at day 0, 105 cells from each cell population were seeded in MegaCell medium. At different time points, cells were counted in triplicates using a hemocytometer and split according to their confluency. For more details about culturing, see Cell Culture section. Wt or GRMD Ven CPCs were grown in DMEM 20% FBS without any cytokines. Two days after culture, media were analyzed for bFGF and vascular endothelial growth factor (VEGF) secretion using the Human FGF-Basic and VEGF Mini ELISA Development Kit from Peprotech according to the manufacturer's protocols (Peprotech, Rocky Hill, NJ, USA). Plates were analyzed using the Model 680 Microplate Reader (BioRad, Herts, UK).
Reagents
Antibodies used in the study are listed in Table 1. Nuclei were stained with DAPI from Sigma (Sigma-Aldrich, St. Louis, MO, USA). For endothelial differentiation, fluorescent-labeled human acetylated low-density lipoprotein (acLDL) was purchased from BTI (Biomedical Technology, Inc., Stoughton, MA, USA). Neonatal canine heart extracts were purchased from Gentaur (Kampenhout, Belgium.).
List of Antibodies Used

Plasmid Cloning and Expansion
Plasmids were prepared using standard procedures. Bacterial stocks were grown in Luria-Broth medium overnight at 37°C. All plasmid preparations were obtained using a GenElute™ HP Endotoxin-Free Plasmid Maxiprep Kit (Sigma) and were quantified using a Nanodrop system. pCMV-SPORT6 plasmids carrying hNkx2.5 cDNAs (Clone ID 5225103) were purchased from Open Biosystem (Huntsville, AL, USA) and used for transfection experiments. Total RNA from wt and GRMD CPCs was extracted using a PureLink RNA mini kit (Invitrogen, Carlsbad, CA, USA), and genomic DNA was digested using the TURBO DNA-free kit (Ambion). cDNA was synthesized using superscript reverse transcriptase III (Invitrogen, Carlsbad, CA, USA). For RT-PCR, cDNA was amplified using Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA). For qPCR analysis, Fast SYBR® Green Master Mix was diluted with cDNA and primers (final concentration, 0.5 mM). For cardiac differentiation experiments, qPCR analysis were performed using the primers listed in Table 2. For myogenic differentiation 1 (MyoD1) and myogenic factor 5 (Myf5) expression, primer sets listed below were used for qPCR analysis:
Primer Sequences Used for Species-Specific Identification of Canine Transcripts by qPCR in Differentiation Experiments
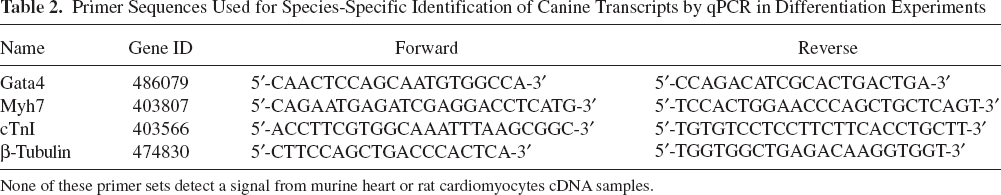
None of these primer sets detect a signal from murine heart or rat cardiomyocytes cDNA samples.
MyoD1 (Gene ID 611940)
Fw 5′-AGCAAAGTCAACGAGGCTTTCGAG-3′
Rv 5′-ACCTTCGATGTAGCGAATCGCGTTG-3′
Myf5 (Gene ID 482582)
Fw 5′-CTACTGTCCTGATGGGCCAAATGT-3′
Rv 5′-GTTGCTCGGAGTTGGTGATTCGAT-3′
Small RNAs were extracted from cell cultures following differentiation using a mirVana kit (Ambion, Austin, TX, USA). miRNAs were retrotranscribed using TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystem, Foster City, CA, USA) with a specific probe annealing the miRNA of interest. Taqman qPCR was performed using TaqMan® Universal PCR Master Mix, No AmpErase® UNG (Applied Biosystem, Foster City, CA, USA) with specific probe sets.
In Vitro Differentiation
For cardiac differentiation assays, starvation experiments were performed by shifting cells from MegaCell growth medium to DMEM with 2% horse serum for 14 days. Differentiation with chemical treatment was induced by treating cells with 10 μM 5′-azacytidine (Sigma-Aldrich, St. Louis, MO, USA) in differentiation medium for 14 days. For human Nkx2.5 overexpression experiments, 24 h after transfection, cells were serum-starved and cultured for 14 days. A coculture protocol was also tested for induction of cardiac differentiation in adult canine CPCs using rat neonatal cardiomyocytes (CMs). Cardiac CPCs and CMs were cultured at a 1:5 ratio, and cultures were shifted to differentiation medium (DMEM supplemented with 2% horse serum). In this case, 7-day cocultures were fixed and stained with antibodies against myosin (MF20) and lamin A/C. Nuclei were counterstained with DAPI. In case of proliferating experiments CPCs/CMs cocultures were kept in DMEM with 10% FBS for 2 days and then fixed for immunocytochemistry experiments. Transforming growth factor-β (TGF-β treatment (1 ng/ml, Sigma-Aldrich, St. Louis, MO, USA) for 7 days was used for smooth muscle induction. Adipogenic and osteogenic assays were performed using mesenchymal adipogenesis or osteogenesis kit from Chemicon (Millipore, MA, USA). For osteogenic differentiation, hBMP2 (human bone morphogenetic protein; 50 ng/ml, Sigma-Aldrich, St. Louis, MO, USA) was added. Briefly, 1 × 105 CPCs were seeded in six-well dishes and cultivated in conditioned media for 1 month according to the manufacturer's protocol. At differentiation endpoints, cells were stained with Red Oil O or Alizarin Red to assay adipogenic or osteogenic differentiation, respectively. For endothelial commitment, 5 × 105 were cultured in ECMatrix™ from the in vitro angiogenesis assay kit according to the manufacturer's protocol (Chemicon Millipore, MA, USA) and stained for endothelial commitment. For skeletal muscle differentiation, wt or GRMD Ven CPCs were cocultured in a 1:5 ratio with C2C12 myoblasts and cultured in serum starvation conditions.
In Vivo Experiment
Severe combined immunodeficient (SCID)/Beige mice and CD1 pregnant rats were purchased from Charles River Laboratories (Wilmington, MA, USA). SCID/Beige mice (10 weeks old) were time-mated to obtain neonates of known gestational age. All animal procedures were carried out under “Biosafety Level 2” conditions approved by the Biosafety Committee of the Katholieke Universiteit Leuven and conform to Directive 2010/63/EU of the European Parliament. All neonatal injections were performed within the first 24–36 h of life. The mother of each litter (singly housed) was removed from the cage, and pups were placed briefly on a bed of ice for anesthesia. A light source was used to visualize thoracic area and the 20-μl volume containing 2.5 × 105 Ven CPCs was administered slowly intraventricularly using a 33-gauge needle and syringe by subxiphoid approach. For identification of injected animals from their noninjected littermates, specific areas of each injected pup was marked by subcutaneous injection of approximately 1 μl of colloidal carbon suspension immediately following stem cell administration. For golden retrievers carrying the GRMD mutation, research was performed according to the principles outlined in the Guide for Laboratory Animal Facilities and Care. Research protocols and facilities were approved by the Animal Care and Use Committee of the Veterinary School of Alfort (Paris, France). For cardiac biopsy isolation, all dogs were maintained until natural death, at which time deceased animals underwent necropsy.
Statistical Analysis
Values are expressed as mean ± SEM. When two groups were compared, an unpaired Student's t test was used. A probability of less than 5% (p < 0.05) was considered to be statistically significant. When three groups were compared and a two-way ANOVA was used, a probability of less than 5% (p < 0.05) was considered to be statistically significant (Bonferroni posttest). Statistical significance of the differences between the percentage values was assessed by the Kruskal–Wallis one-way analysis of variance (ANOVA) on ranks, with a Dunn post hoc test. a represents the significance in each independent case. α < 0.05 was considered statistically significant.
Results
In Vitro Isolation and Phenotypic Characterization
Heart biopsies obtained from different regions (atrium, aorta, and ventricles) of wt and GRMD dogs (Fig. 1A, B) were cut in small pieces and plated in 1% gelatin or collagen coated dishes (50). Donor age ranged from 12 to 18 months for wt dogs, while GRMD dogs were, at maximum, 6 months old (Table 3) (16). At 7–10 days, bright round cells loosely adhering upon a fibroblast feeder layer (Fig. 1C, D) were collected and expanded in MegaCell medium. Cell outgrowth was observed from biopsies originating from the aorta (Ao), atrium (At), and ventricular (Ven). Collagen and gelatin coating promoted cell outgrowth at similar levels (data not shown). Floating cells were successfully cloned by serial dilution, and different clones from wt and GRMD biopsies were further characterized in terms of proliferative ability and gene profile expression. At low density, cells acquired homogenous spindle morphology and required a gelatin coating for complete adhesion to plastic dishes (Fig. 1E, F). After 3 or 4 days, cell clone cultures became confluent and required passaging, as indicated in the methods section (Fig. 1G, H). Initially, these clones were characterized for the expression of AP as pericyte marker. AP signal was also detected from outgrowing cells in fresh explants of wt and GRMD biopsies (Fig. 1I, J). In both low (Fig. 1K, L) and high (Fig. 1M, N) density cultures, more than 70% of cells were positive for AP; however, there were no detectable differences between wt and GRMD cells.

Morphological characterization of canine cardiac progenitors isolated from different heart regions. (A, B) DAB staining for dystrophin expression in cardiac tissue sections from wild type (wt) (A) and Golden Retriever muscular dystrophy (GRMD) (B) dogs. Scale bars: 100 μm. (C–H) Phase contrast (PhCo) images of cellular outgrowth from wt (C) and GRMD (D) cardiac biopsies showed small bright cells underlying a layer of fibroblast feeder, representing putative cardiac progenitors (CPCs). Middle and bottom: PhCo images of growing (E, F) and confluent (G, H) wt and GRMD CPCs. (I–N) Alkaline phosphatase (AP) staining on outgrowing cells from wt (I) and GRMD (J) cardiac explants. At midconfluency (K, L) and full confluency (M, N) stages, the bulk population contains a pool of AP+ cells (about 60–70%) that are maintained during culturing. Scale bars: 100 μm (for C–D and I–J) and 50 μm (for E–H and K–N). Data presented are from three independent experiments.
Characteristics of the Animals Used for CPCs Isolation

Three healthy control biopsies were obtained from dogs died for natural causes not related with cardiac dysfunction. Three cardiac biopsies were obtained from animals affected by GRMD.
Characterization of Surface Markers and Gene Expression Profile
Adult cell clones were analyzed by RT-PCR for the expression of early cardiac and mesodermal markers. RNA was extracted from wt and GRMD clones derived from At, Ao, and Ven regions. Notably, four clones per cardiac region were characterized from individual wt and GRMD dogs with a very high degree of homogeneity in terms of marker expression profile between clones of the same region (Tables 4 and 5). A strong network of cardiac gene expression has been observed in wt and to a lesser extent in GRMD clones. As previously reported (12,60), early cardiac markers such as Gata4, Gata6, and heart and neural crest derivatives expressed 2 (Hand2) mRNAs are expressed by both proliferating wt and GRMD Ven clones (Tables 4 and 5). Furthermore, wt and GRMD Ven clones expressed the transcription factors myocyte enhancer factor 2A (Mef2A) and Mef2C that are involved in myocyte differentiation and stress signaling (6,35). Previous studies reported the early mesodermal genes Flk1 and cKit act as markers of cardiac progenitor cells (5). Both Flk1 (fetal liver kinase, also known as VEGF receptor 2) and cKit were expressed in our Ao- and Ven-derived clones (Tables 4 and 5). Endogenous Nkx2.5 expression has never been detected in proliferating clones (both at mRNA and protein levels) independently from pathological conditions. In subconfluent cultures, wt and GRMD Ven clones expressed the pericyte markers platelet-derived growth factor receptor a (PDGFRα), PDGFRβ, chondroitin sulfate proteoglycan 4 (NG2), a smooth muscle actin (αSMA), as detected by Western blotting and immunofluorescence analysis (Fig. 2A–D). Wt and GRMD clones also homogenously expressed GATA4 at the protein level, as detected by immunofluorescence (IF) analysis (Fig. 2E, F). Interestingly, serial dilution cloning allows the removal of endothelial, smooth muscle cells and fibroblast contaminations, detected only in the polyclonal populations but not in the corresponding clones (Table 6). Due to their wide range of early cardiac and pericyte marker expression, we focus our analysis on the ventricle-derived clones of wt and GRMD animals. Nonetheless, considering the similarities in terms of cell morphology and gene expression profile to cardiac progenitor cells as previously reported (1,4,20,29,31,41), we operationally termed our cells “canine cardiac progenitor cells” (CPCs).
Gene Expression Profile Analysis of Proliferating wt CPCs Clones

r, RT-PCR; p, WB; if, immunofluorescence; f, FACS.
Gene Expression Profile Analysis of Proliferating GRMD CPCs Clones
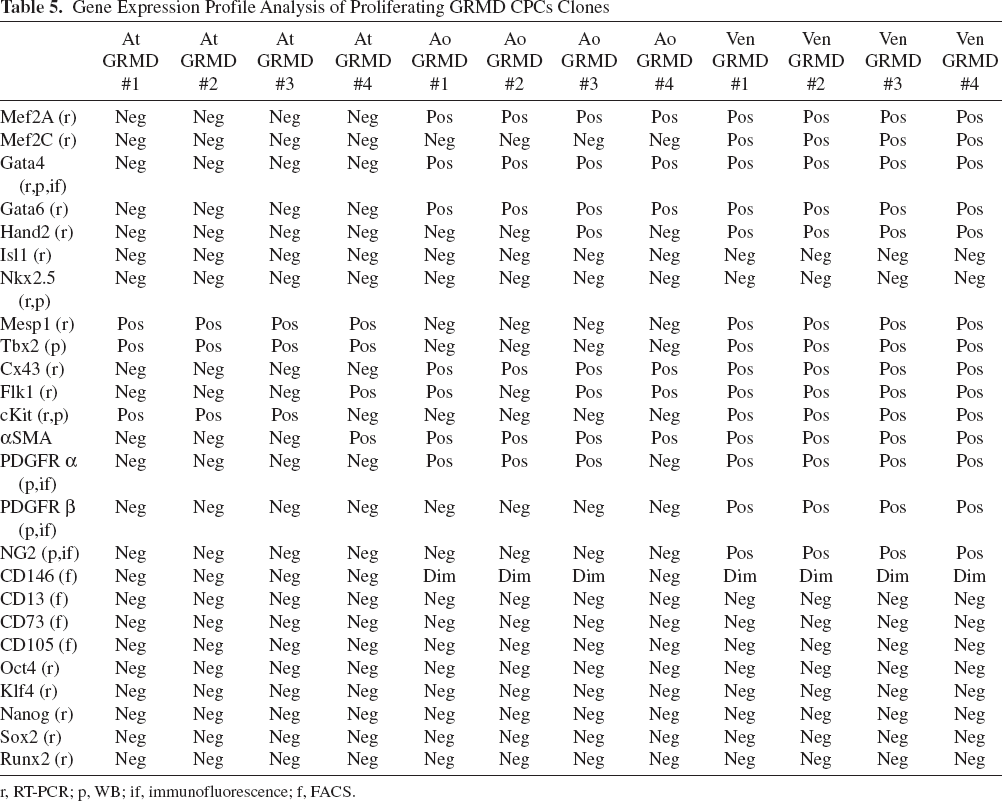
r, RT-PCR; p, WB; if, immunofluorescence; f, FACS.
Heterogeneity at Gene Expression Level Between Polyclonal and Clonal Populations of wt and GRMD CPCs

r, RT-PCR; p, WB; if, immunofluorescence; f, FACS.
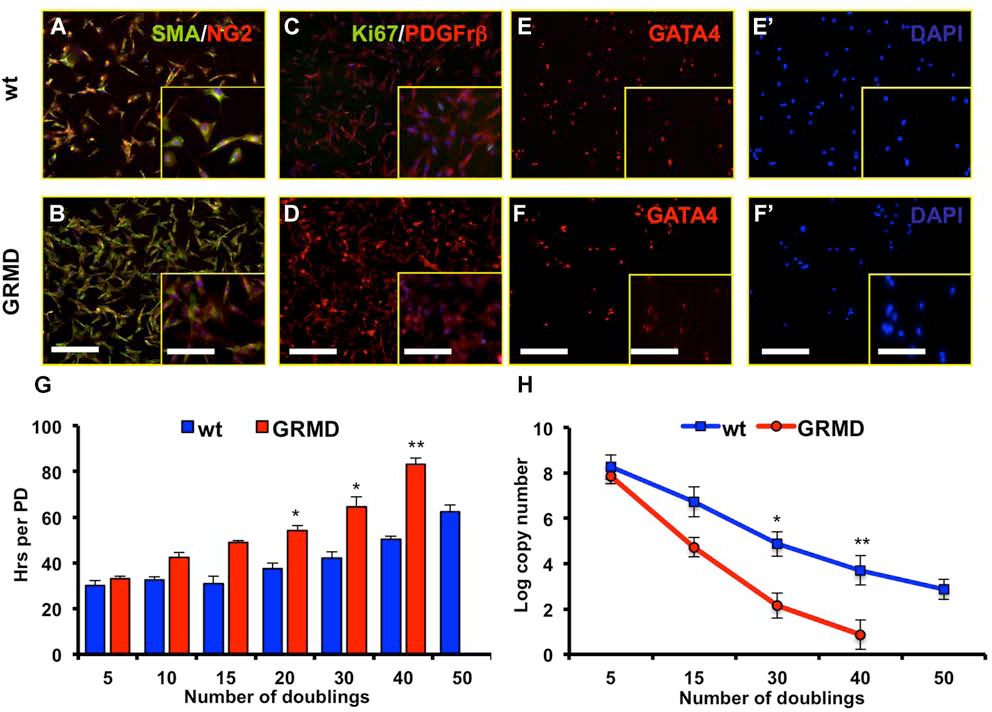
Markers expression profile of CPCs. (A, B) Immunofluorescence (IF) analysis showing αSMA (green) and NG2 (red) expression in wt (A) and GRMD (B) ventricle (Ven) CPCs. Nuclei were counterstained in blue with DAPI. Scale bar: 100 μm. Inset scale bar: 50 μm. Data presented are from three independent experiments. (C, D) IF analysis revealing PDGFRβ+ (red)/Ki67+ (green) cells in wt (C) and GRMD (D) Ven CPCs. Nuclei were counterstained in blue with DAPI. Scale bar: 100 μm. Inset scale bar: 50 μm. Data displayed are representative of three independent experiments. (E, F) IF analysis showing GATA4 (red) expression in wt (E) and GRMD (F) Ven CPCs. Nuclei were counterstained in blue with DAPI in E′ and F′. Scale bar: 100 μm. Inset scale bar: 50 μm. Data presented are from three independent experiments. (G) PD analysis of freshly isolated wt (blue bars) and GRMD (red bars) Ven CPCs. The proliferative gap increased during time and became evident at 15 doublings. Values are presented as means ± SEM (n = 5, *p < 0.05 and **p < 0.01 wt vs. GRMD). (H) Telomeres copy number quantification of wt (blue line) and GRMD (red line) Ven CPCs. Values from atrium (At), aorta (Ao), and Ven clonal populations are presented as means ± SEM (n = 5, *p < 0.05 wt vs. GRMD and **p < 0.01 wt vs. GRMD).
Proliferative Capacity and Telomerase Assay
Proliferative capacity of wt and GRMD ventricle clones was examined by monitoring PD and by measuring telomerase activity. Considerable differences were observed between wt and GRMD Ven CPCs. On average, wt cells were expanded for more than 50 PD in culture whereas GRMD CPCs underwent senescence earlier (40 PD), showing a limited lifespan. Senescent cells can be easily identified as they acquire a distinct fibroblastic shape with a flat, spindle morphology. PD analysis was consistent with these observations with GRMD Ven CPCs displaying a slower proliferation rate at all the PD analyzed (Fig. 2G). After 15 PD, GRMD Ven CPCs showed a slower proliferation rate compared with wt cells, differing by 18 h (Fig. 2G, compare blue with red bars). By contrast, wt cells exhibited a prolonged lifespan and faster proliferation rate (average doubling time 31 h) compared with GRMD Ven CPCs. This impairment of proliferation was more evident at later doublings where GRMD Ven CPCs reached senescence and wt progenitors were still actively dividing with a doubling time of 62 h. As high telomerase activity is known to sustain cell proliferation and circumvent replicative senescence (21), we examined telomerase activity in wt and GRMD Ven CPCs and calculated the log copy number of telomeric sequences at different doublings. In agreement with previous observations, telomeric copy number variation encompassed the proliferative defect of GRMD progenitors. Telomerase activity of GRMD Ven CPCs dramatically declined after 15 doublings and became barely detectable after 40 PD (Fig. 2H, red line). In contrast, telomerase levels in wt Ven CPCs declined gradually but were still detectable after 40 doublings when GRMD cells had ceased replicating (Fig. 2H, compare blue with red lines). Telomerase levels in wt Ven CPCs were significantly decreased only after 50 doublings, supporting the results obtained from proliferation analysis.
Mesodermal Differentiation Potential
To assess the in vitro potential of Ven CPCs to convert into mesodermal cell types, subconfluent clones at various PD were cultured in media known to induce specific cell differentiation. During the proliferative phase, both wt and GRMD Ven CPCs lacked expression of key regulators of myogenic commitment, such as MyoD1 and Myf5 (15) (Fig. 3A). Consequently, Ven CPCs in serum starvation conditions did not spontaneously form myotubes or fuse with C2C12 myoblasts, confirming impairment of skeletal myogenic commitment (Fig. 3B, C). Augmenting the neovascularization in response to coronary heart diseases represents a major challenge for cell-based therapies. Towards this aim, endothelial commitment of healthy and GRMD Ven CPCs was examined by culturing cells in solid three-dimensional scaffolds enriched in various proteins (see In Vitro Differentiation). Three days after seeding, wt and GRMD Ven CPCs rapidly aligned and formed hollow tube-like structures identical to those of differentiated HUVEC cells (Fig. 3D, E). Moreover, IF analysis confirmed the robust endothelial commitment of Ven CPCs showing expression of mature endothelial markers such as CD31 (platelet endothelial cell adhesion molecule; PECAM) (Fig. 3F, G) and acLDL uptake ability (Fig. 3H, I). It is noteworthy that Ven CPCs started to express CD31 only upon differentiation and not during the proliferative state (data not shown). Wt and GRMD Ven CPCs also showed the ability to differentiate into smooth muscle cells when treated with TGF-β. The dystrophic condition did not interfere with smooth muscle differentiation and when stimulated approximately 50% of cells expressed mature smooth muscle markers such as calponin and l-caldesmon (Fig. 3J, K). Furthermore, adult canine CPCs could differentiate into adipocytes (40% of the total population) when cultured under specific conditions. Both wt and GRMD Ven CPCs formed small Red Oil O-stainable lipid droplets into the cytoplasmic compartment (Fig. 3L, M). However, these cells do not express the osteogenic marker Runt-related transcription factor 2 (Runx2) (Tables 4 and 5) and consequentially failed to differentiate into mineralized osteocytes as confirmed by Alizarin Red staining (Fig. 3N, O).

In vitro differentiation of CPC towards mesodermal lineage. (A) qPCR analysis for MyoD1 and Myf5 expression in proliferating wt (blue bars) and GRMD (red bars) Ven CPCs. Proliferating C2C12 were used as a positive control (black bars) (n = 5 *p < 0.01 C2C12 vs. wt **p < 0.01 C2C12 vs. GRMD). (B, C) Cocultures (ratio 1:5) of murine C2C12 with wt (B) or GRMD (C) Ven CPCs stained for lamin A/C (red) and MyHC (green) expression. Nuclei were counterstained in blue with DAPI. Scale bar: 50 μm. Data presented are representative of three independent experiments. (D–I) Phase contrast and IF images of Ven CPCs cultured in three-dimensional ECMatrix™ scaffold for 7 days. Cells rapidly assumed hollow tubular-like structures (PhCo; D, E) and were further characterized for CD31 (PECAM; F and G in green) and acLDL (H and I in red) expression as mature markers of endothelial differentiation. Scale bars: 100 μm. Data presented are representative of three independent experiments. (J, K) One-week stimulation with TGF-β was sufficient to activate smooth muscle program in both wt (J) and GRMD Ven CPCs (K) as highlighted by the presence of cells positive for calponin (green) and caldesmon (red) expression. Nuclei were counterstained in blue with DAPI. Scale bar: 100 μm. Data presented are representative of three independent experiments. (L, M) Red Oil-O staining revealed lipid droplets accumulation both in wt (L) and GRMD progenitors (M) treated for 1 month with adipogenic-conditioned media. Scale bar: 100 μm. Inset scale bar: 50 μm. Data presented are representative of three independent experiments. (N, O) Ven CPCs cultured for 1 month in hBMP2-conditoned media were counterstained for Alizarin Red confirming the absence of calcium deposits both in wt (N) and GRMD (O) progenitors. Scale bar: 100 μm. Inset scale bar: 50 μm. Data presented are representative of three independent experiments.
Cardiac Differentiation
We evaluated the ability of canine CPCs to undergo cardiac differentiation by testing several differentiation protocols. In addition to the classical differentiation protocol of serum starvation with or without addition of 5-azacytidine (5-Aza) (11,25), we also overexpressed hNkx2.5 in these cells by transient transfection. Direct comparison of the differentiation rate between wt and GRMD Ven CPCs was performed in order to highlight the discrepancy in stemness between these cells. Expression of late cardiac markers at differentiation endpoint was examined by immunofluorescence and qPCR analysis, the latter taking advantage of primer sets that specifically recognize canine sequences (Table 2). With these primers, GATA4, cardiac myosin heavy chain (cMyHC), and cardiac Troponin I (cTnI) expression was not detected in mice heart tissue and rat neonatal cardiomyocytes since those primer sets do not recognize the murine or rat version of these genes (data not shown). When proliferating, wt and GRMD Ven CPCs actively expressed GATA4, but no late cardiac markers (Fig. 4A). When Ven CPCs were exposed to cardiac differentiation medium (low serum) they initiated cardiac differentiation (Fig. 4B), to varying extents between wt and GRMD progenitors (compare Fig. 4C and D). In fact, qPCR analysis showed 10-fold upregulation of GATA4 and cMyHC in wt compared with GRMD Ven CPCs (Fig. 4E). Under these conditions, we obtained differentiation rates of 35% and 23% for wt and GRMD progenitors, respectively (Fig. 4B). 5-Aza treatment increased the overall number of differentiated cells (Fig. 4F, G) as confirmed also by qPCR analysis for late differentiation markers (Fig. 4H), resulting in approximately 55% αSA and cTnI positive wt cells and only 30% positive GRMD Ven CPCs. Due to the lack of endogenous Nkx2.5 expression in both proliferating wt and GRMD CPCs (previously mentioned, see Tables 4 and 5), we thought to enhance the cardiac differentiation rate by genetically restoring its expression. Human and canine sequences of Nkx2.5 strongly overlap, with a 95% protein homology (data not shown). Transfection efficiency was comparable between wt and GRMD Ven CPCs as hNkx2.5 expression levels were not significantly different when measured 48 h after transfection (wt ΔΔCt = 1.23 ± 0.43 vs. GRMD DDCt = 1.02 ± 0.11; data not shown). Transient overexpression of human Nkx2.5 (hNkx2.5) resulted in the most efficient cardiac differentiation of all methods tested with 70% αSA+/cTnI+ cells present in wt Ven CPC cultures (Fig. 4I). Under the same conditions, cardiac commitment of GRMD Ven CPCs was lower and comparable with the 5-Aza protocol (about 35%), suggesting that GRMD Ven CPCs are insensitive to hNkx2.5 overexpression (compare Fig. 4I and J). Upon this condition, Ven CPCs tend to form cellular structures comparable with rat neonatal cardiomyocytes (Fig. 4K). The cardiac inductive effect of hNkx2.5 overexpression was confirmed by qPCR analysis, showing robust upregulation of cMyHC and cTnI only in wt Ven CPCs. In fact, cMyHC and cTnI levels in wt Ven CPCs were 15- and 20-fold higher than in GRMD progenitors, respectively (Fig. 4L). As documented by Western blot analysis, only terminally differentiated wt Ven CPCs can express dystrophin as a hallmark of cardiomyogenesis contribution. As expected, the GRMD Ven counterpart failed to express the structural protein (Fig. 4M).
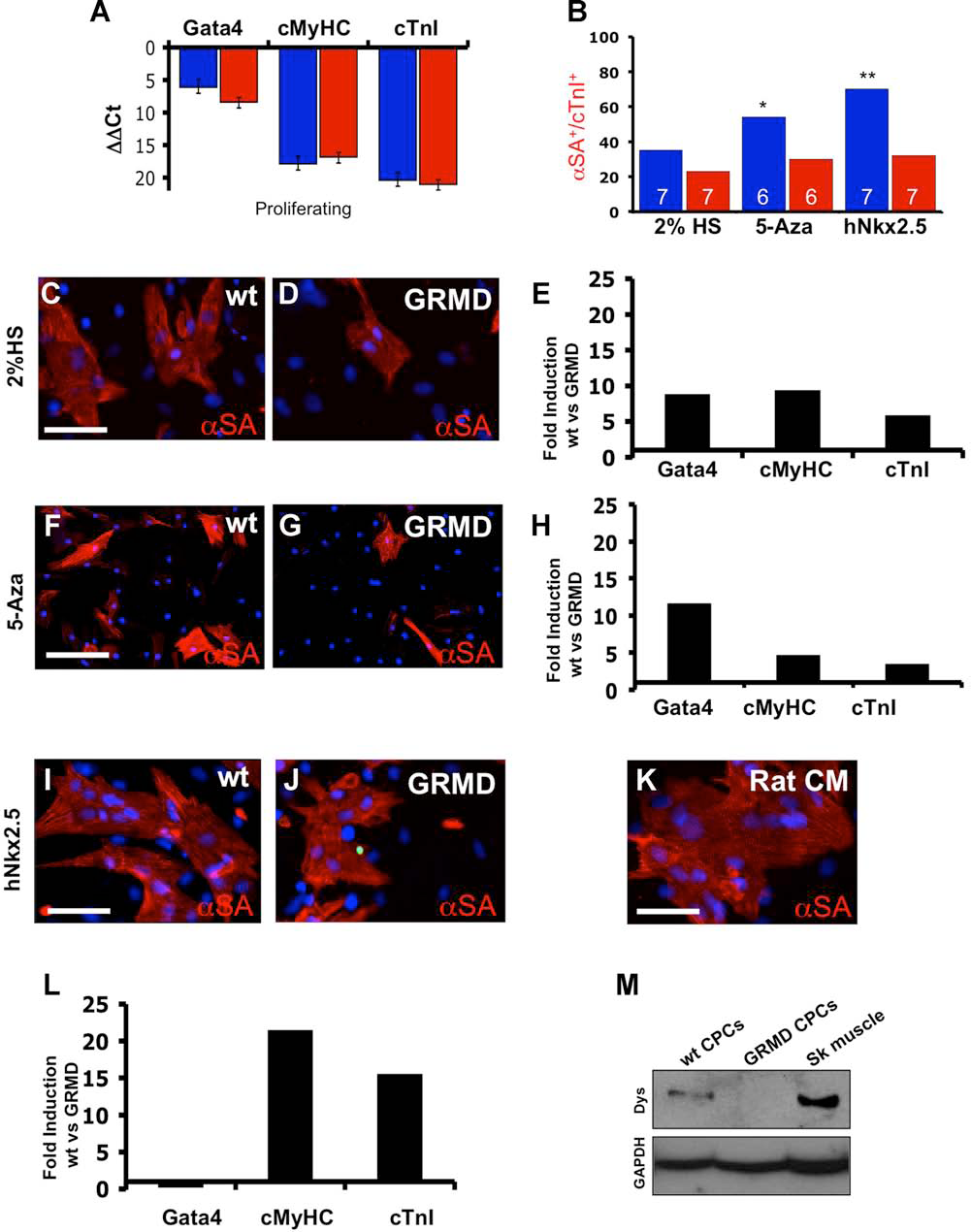
Cardiac induction of CPCs. (A) qPCR analysis for early and late cardiac markers in proliferating wt (blue bars) and GRMD (red bars) Ven CPCs (n = 3). (B) Cardiac differentiation rates of wt (blue bars) and GRMD (red bars) Ven CPC in three differentiation protocols. Cardiac commitment rate was calculated as the ratio of double αSA+/cTnI+ cells versus the total number of cells. Numbers of experiments performed are indicated within the bars. *α < 0.05, **α < 0.01. (C, D) Cardiac differentiation of wt (C) and GRMD (D) Ven CPC induced by serum starvation. After 14 days, cells were analyzed for the expression of αSA (red) as marker of terminal differentiation. Serum starvation condition activates αSA expression (red) in differentiated cells. Note that number of αSA+ cells resulted always higher in wt than in GRMD CPC (compare C with D). Nuclei were counterstained with DAPI. Scale bar: 50 μm. Data displayed are representative of seven independent experiments. (E) Fold induction rate relative to Gata4, cMyHC, and cTnI expression of wt versus GRMD progenitors in serum starvation conditions. Expression levels were compared at differentiation endpoint (n = 7). (F, G) IF staining for αSA (red) expression of wt (F) and GRMD (G) Ven CPCs following 5-azacytidine treatment. Nuclei were counterstained in blue with DAPI. Scale bar: 100 μm. Data displayed are representative of five independent experiments. (H) Fold induction rates of Gata4, cMyHC, and cTnI expression of wt versus dystrophic progenitors at differentiation endpoint in 5-azacytidine treatment (n = 5). (I, J) Cardiac differentiation analysis of wt (I) and GRMD (J) Ven CPC induced by forced hNkx2.5 expression. After 14 days, cells were analyzed for the expression of αSA (red) as marker of terminal differentiation. Forced hNkx2.5 expression markedly upregulates αSA expression (red) in differentiated cells. Note that number of αSA+ cells resulted always higher in wt than in GRMD CPC (compare I with J). Nuclei were counterstained with DAPI. Scale bar: 50 μm. Data displayed are representative of seven independent experiments. (K) IF staining for αSA (red) on primary culture of rat neonatal cardiomyocytes. Nuclei were counterstained with DAPI. Scale bar: 50 μm. (L) Fold induction rate relative to Gata4, cMyHC, and cTnI expression of wt versus GRMD progenitors following hNkx2.5 overexpression. Expression levels were compared at differentiation endpoint (n = 7). (M) Western blot (WB) analysis for dystrophin expression from wt and GRMD Ven CPCs at differentiation endpoints. Canine skeletal muscle as a positive control. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as internal control (n = 3).
miRNA Profiling During Cardiac Differentiation
We further analyzed the microRNA (miRNA) expression profile during cardiac differentiation, focusing our attention on miRNAs involved in cardiomyogenic and fibrotic events (8,33). miR-208, miR-1, and miR-133a are involved in cardiac commitment and their expression is regulated by αMyHC and MEF2A, respectively (9,24,55). miR-21 is known to regulate the extent of interstitial fibrosis and cardiac hypertrophy (49), whereas miR-206 expression is restricted to the skeletal muscle system (26). miRNA expression levels were compared between wt and GRMD Ven CPCs during proliferation, serum starvation, and transient hNkx2.5 overexpression. Myo-miR-206 expression was never detected in wt and GRMD CPCs confirming the lack of skeletal muscle commitment (data not shown). During the proliferative state, expression of miR-133a and miR-1 was slightly higher in wt than in GRMD Ven CPCs (Fig. 5A, white bars) whereas miR-208 expression was barely detectable in both CPCs. However, serum starvation (Fig. 5A, red bars) and forced hNkx2.5 expression (Fig. 5A, blue bars) strongly activated miR-133a, miR-1, and miR-208 expression only in wt Ven CPCs. In the case of hNkx2.5 overexpression, myosin-dependent miR-208, miR-133a, and miR-1 were robustly overexpressed (9- to 10-fold) in wt compared with GRMD Ven CPCs. Interestingly, GRMD Ven CPCs had markedly higher miR-21 expression than wt in all the protocols tested, with levels ranging from 5- to 10-fold higher (Fig. 5A). This miRNA expression profile thus confirms the greater cardiac commitment of wt Ven CPCs and further highlights the diminished plasticity of GRMD Ven CPCs towards cardiomyogenic fate.

microRNA profiling and paracrine release of growth factors. (A) Relative expression of miR-133a, miR-208, miR-1, and miR-21 of wt versus GRMD Ven CPCs. miRNAs fold induction rate was analyzed at different timepoints: during proliferation (white bars), differentiation endpoints in serum starvation (red bars) and hNkx2.5 overexpression (blue bars). All the experiments were performed in triplicates per each clone. (B, C) IF staining for GFP (green) and Ki67 (red) of rat neonatal cardiomyocytes cocultured for 2 days with GFP+ wt (B) and GRMD (C) Ven CPCs. Nuclei were counterstained in blue with DAPI. Scale bar: 100 μm. Data are representative of four independent experiments. (D) Rate of cardiomyocytes proliferation was calculated as the ratio of Ki67+/GFP- cells versus the total number of GFP- cells. Proliferating rate was analyzed in coculture conditions with wt (blue bar) or GRMD (red bar) Ven CPCs. Gray bar (CM) represents proliferating rate of rat neonatal cardiomyocytes alone. Values are presented as means ± SEM (n = 4 * p < 0.01 CM + wt vs. CM + GRMD; **p < 0.01 CM + wt vs. CM alone). (E) bFGF and VEGF secretion in proliferating media of wt (blue bars) and GRMD (red bars) Ven CPCs quantified by ELISA assay. Values are presented as means ± SEM (n = 4). (F) qPCR analysis in growing conditions revealed robust expression of cMet, HGF, and IGFR-1 in wt (blue bars) and GRMD (red bars) Ven CPCs. Data presented are from five independent experiments ± SEM.
Paracrine Effects of CPCs
A key advantage of cell transplantation compared with protein or gene therapy is that donor cells may also alleviate chronic heart dysfunction by stably releasing angiogenic and prosurvival factors (28,30). Moreover, cell–cell contacts or secretion of humoral factors may facilitate the dedifferentiation process and cell cycle re-entry of endogenous cardiomyocytes, an event frequently coupled with cardiac regeneration in newt and zebrafish and that could support cardiac healing also in mammals during post-natal life (42,58). To test this hypothesis, we transduced wt and GRMD Ven CPCs with a GFP-expressing lentiviral vector. FACS-based sorting was performed to enrich the GFP+ fraction of wt and GRMD Ven CPCs. Rat neonatal cardiomyocytes (99% pure population) were cocultured together with GFP+ Ven CPCs to determine to what extent rat cardiomyocyte dedifferentiation occurred. Ki67 reactivity was observed in many GFPnegative cells, representing actively proliferating rat cardiomyocytes (Fig. 5B, C). In this condition, 18% ± 2.3% and 11.3% ± 0.7% of primary rat cardiomyocytes were Ki67 positive when cocultured with wt or GRMD Ven CPCs, respectively (Fig. 5D, blue and red bars). In our hands the dedifferentiation rate of rat cardiomyocytes alone was 2% ± 1% (Fig. 5D, gray bar). Next we aimed to establish whether our cells could potentially act as paracrine mediators of cell survival and increase angiogenesis. Proangiogenic factors, such as VEGF and bFGF, were measured by ELISA assay in the media of proliferating wt and GRMD CPCs. Indeed, we found similar expression levels of both growth factors by wt and GRMD Ven CPCs (Fig. 5E). The activation of hepatocyte growth factor (HGF) and insulin-like growth factor-1 (IGF-1) networks provides a selective advantage to cardiac progenitors enabling them to counteract post-ischemic cardiac degeneration (31). The HGF-cMet system is implicated in cell migration and to a lesser extent, in cell growth and survival, whereas IGF-1/ IGFR-1 has a predominant effect on cell multiplication and viability (7,57). For this reason, we analyzed the expression of these factors in our cells by qPCR. Both wt and GRMD Ven CPCs expressed high levels of cMet, HGF, and IGFR-1 during their proliferative state (Fig. 5F). Altogether these results indicate that Ven CPCs may activate paracrine signaling in acute or chronic cardiac dysfunctions by promoting antiapoptotic and proangiogenic effects.
In Vivo Regeneration Potential
To further investigate cardiac commitment of wt and GRMD Ven CPCs, we tested their in vivo ability to participate in cardiomyogenesis and colonize hearts of neonatal immunodeficient mice. It has been recently reported that the mammalian heart appears to have a great plasticity within a brief period after birth (42). Due to this strong regenerative capacity, the 1-day-old mouse heart can be considered as a useful model to explore the in vivo cardiomyogenic potential of our Ven CPCs. We took advantage of the piggy-bac transposon system to genetically label CPCs with nuclear red fluorescent protein (dsRed) to allow an easy and precise in vivo identification of canine integrated cells in the host myocardium (Fig. 6A). To our knowledge, canine genome editing by transposon technology has never been described in the literature nor has it been used for marking CPCs. Therefore, our approach represents the first attempt to use this novel nonviral technology in cardiac cell therapy and in vivo tracing. Nuclear tagging enables easy identification of transplanted cells, allowing for better discrimination of fusion events between donor cells and resident cardiomyocytes. In vivo experiments were performed with Ven CPCs clones due to their higher cardiac differentiation potency, as previously demonstrated in vitro. Following double transfection with transposase (TS)- and transposon (TN)-carrying plasmids, approximately 25% of the cells were positive for nuclear dsRed expression (Fig. 6B, C). Although only 0.5–1% of the cells stably expressed the dsRed transposon element after a few doublings (Fig. 6D, E), they could be enriched by FACS to achieve up to 99% dsRed+ cellular fractions (Fig. 6F, G). No striking differences were observed in terms of transposon integration between wt and GRMD Ven CPCs (Fig. 6H, I). Sorted cells maintained the morphology and proliferation ability of nonlabeled cells and were easily expanded to a suitable number for transplantation experiments in a large animal model (Fig. 7A, B). Moreover, dsRed+ CPCs retained the ability to differentiate into cardiomyocyte-like cells, as demonstrated by late cardiac markers expression in serum starvation conditions (Fig. 7C–F). 2.5 × 105 dsRed+ wt or GRMD Ven CPCs were injected directly in the cardiac wall of 1-day-old SCID/ Beige pups (Fig. 8A). As a negative control, we included a cohort of mice treated with an equal cell number of dsRed+ wt canine skin fibroblasts. At 1-month postinjection, we analyzed CPCs integration in the host cardiac system by immunolabeling with dsRed and αSA antibodies. All the animals survived the experimental procedures except for one animal that died within 24 h of cell injection. At the time of dissection, all transplanted hearts appeared morphologically and functionally normal (data not shown). As shown in Figure 8, wt and GRMD Ven CPCs colonize the cardiac system by forming new αSA+ cardiomyocytes (Fig. 8B, C). Nuclear dsRed expression was detected also in cardiac cells positive for myosin heavy chain (Fig. 8D, E), confirming the cardiomyogenic fate of wt and GRMD Ven CPCs. As expected, only wt Ven CPCs contributed to dystrophin expression in the host myocardium (Fig. 8F) whereas GRMD CPCs failed to express this protein (Fig. 8G). As a further confirm of their cardiomyogenic potential, we detected dsRed+/Cx43+ cells in both mice treated with wt or GRMD Ven CPCs (Fig. 8H, I). To confirm that dsRed signal was specific for exogenous canine cells, nuclear colocalization of dsRed and lamin A/C expression were examined in both wt and GRMD Ven CPCs (Fig. 8J, K). However, GRMD Ven CPCs contributed to cardiomyogenesis to a lower extent than the wt counterpart, as revealed by percentage quantification of double MyHC+/dsRed+ cells (24.7% ± 4.34% wt vs. 14.44% ± 3.64% GRMD; yellow fractions in circle graphs in Fig. 8L). In vivo transplantation of GRMD Ven CPCs augments the pool of MyHC-/dsRed+ cells (2.11% ± 1.02% wt vs. 4.41% ± 0.87% GRMD; red fractions in circle graphs in Fig. 8L), which most likely corresponds to interstitial cells. It is important to mention that the quantitative analysis has been performed only on tissue sections corresponding to regions proximal to the injection site. When considering the total amount of cells engrafted per single heart, 1,875 ± 15 wt Ven CPCs were counted in spite of the 1,075 ± 27 cells for the GRMD counterpart. Within the pool of engrafted cells, wt Ven CPCs contributed to the higher number of MyHC+ cells per heart (1,699 ± 45) compared with the GRMD counterpart (717 ± 52) (yellow bars in Fig. 8L). Conversely, the pool of interstitial cells (MyHC-) was increased following GRMD Ven CPCs transplantation (336 ± 21 GRMD vs. 176 ± 33 wt) (red bars in Fig. 8L). As expected, injection of canine skin fibroblast increased the percentage of interstitial cells (12.83% ± 3.87%) without any contribution to newly formed cardiomyocytes. Altogether, these data are consistent with the previous observation for differentiation experiments in vitro, highlighting the reduced cardiac potential of CPCs derived from a pathological environment.

Genetic labeling and dsRed+ CPC enrichment. (A) Schematic representation of transposase plasmid pcDNA3CMV/bGI-PiggyBAC/MTA (left) and transposon plasmid pBac/CMV-ndsRed (right) used for genetic labeling of wt and GRMD Ven CPCs. (B–G) Following transfection with the transposase/transposon plasmids, wt (B) and GRMD (C) Ven CPCs were analyzed by IF for nuclear dsRed expression (red). After few PD the pool of wt (D) and GRMD (E) dsRed+ CPC decreased due to the progressive clean up of transiently transfected cells without transposon integration. Stable integrating dsRed+ cells were FACS-sorted to obtain a pure bulk of nuclear dsRed+ wt (F) and GRMD (G) Ven CPCs. Scale bars: 100 μm. Data displayed are representative of three independent transfection experiments per each clone. (H, I) FACS-based sorting of dsRed+ wt (H) and GRMD (I) Ven CPCs. P1 gates represent the pool of dsRed+ cells (n = 3).

Amplification and differentiation capacity of dsRed+ CPC. (A) Cell amplification graph of FACS-sorted dsRed+ Ven wt (blue line) and GRMD (red line) CPC. Data displayed are representative of five independent experiments (*p < 0.01 wt vs. GRMD). (B) Population doubling (PD) analysis of FACS-sorted dsRed+ Ven wt (blue line) and GRMD (red line) CPCs. Transposon integration and cell sorting do not interfere with proliferative ability of dsRed+ progenitors that strongly overlaps with non-labeled CPCs (*p < 0.01 wt vs. GRMD). (C–F) IF staining for αSA (green) and dsRed (red) expression on FACS-sorted wt (C–D′) and GRMD (E–F′) Ven CPCs in starvation condition. Nuclei were counterstained in blue with DAPI. White scale bars: 50 μm. Yellow scale bars: 25 μm. Data displayed are representative of four independent experiments.

In vivo transplantation of dsRed+ CPC in neonatal SCID/Beige mice. (A) Technical scheme for in vivo transplantation of CPCs in immunodeficient 1-day-old pups. 2.5 × 105 dsRed+ wt or GRMD Ven CPCs were injected in the cardiac wall of 1-day-old SCID/beige pups. Transplanted hearts were collected at 30 days postinjection for the detection of cardiac chimerism of donor cells in the host myocardium. (B–G) IF staining for αSA (green in B and C), MyHC (green in D and E), dystrophin (green in F and G), and nuclear dsRed (red) expression in 30 days old SCID/Beige hearts injected with wt (B, D, and F) or GRMD (C, E, and G) Ven CPCs. Scale bars: 100 μm. Twenty slides were analyzed per each mice transplanted. (H, I) IF staining for connexin 43 (green in H and I) and nuclear dsRed (red) expression on cardiac tissue sections from mice treated with wt (H) and GRMD (I) Ven CPCs. PhCo images are in H′ and I′. Scale bar: 100 μm. (J, K) IF staining on cardiac sections of SCID/Beige mice treated with wt (J) or GRMD (K) Ven CPCs revealing colocalization of nuclear dsRed (red) and lamin A/C (green) signals in canine transplanted cells. Scale bars: 30 μm. Nuclei were counterstained in blue with Dapi. 20 slides were analyzed per each mice transplanted. (L) Upper circle graphs: percentage quantification of integrating CPCs at 1 month posttransplantation. Only slides containing dsRed+ cells, most likely corresponding to regions proximal to the injection site, were considered for the analysis. Counted cells were divided in three groups: resident murine cardiomyocytes (green bars), CPCs-derived cardiomyocytes (yellow bars), and interstitial CPCs-derived cells (red bars). Canine skin fibroblasts (Fibs) were used as negative controls. Lower bar graphs: cells quantification of canine-derived cardiomyocytes (yellow bars) or interstitial cells (red bars) per single heart treated with wt, GRMD Ven CPCs, or canine skin fibroblasts. Seven mice were analyzed for each group. Values are presented as means ± SEM (*p < 0.01 MyHC+/dsRed+ wt vs. MyHC+/dsRed+ GRMD; ** p < 0.05 MyHC+/dsRed+ wt vs. MyHC-/dsRed+ GRMD).
Discussion
In the present study, we show that the hearts of wt and GRMD dogs have a pool of progenitor cells (CPCs). We demonstrate that resident canine CPCs can be isolated and genetically manipulated and are self-renewing, clonogenic, and paucipotent. However, wt and GRMD Ven CPCs reveal relevant differences in terms of expansion and differentiation ability. While wt cells can be expanded for more than 50 doublings in culture, GRMD CPCs displayed limited lifespan and underwent senescence prematurely at 40 doublings. Senescent cells can be easily identified as they acquire a marked fibroblastic shape with a flat, spindle morphology. Together with these observations, PD analysis highlighted the lower proliferative capacity of GRMD CPCs at all the cell doublings analyzed. These results are consistent with new studies reporting that intrinsic characteristics, such as telomere attrition, are critical to tissue regeneration (44). Considering the broader scientific literature (1,4,20,29,31,41) and the similarities found in terms of marker expression profile with reports from other groups isolating cardiac stem cells, we introduced the term CPCs to indicate cells with cardiogenic commitment. In fact, ventricle clones robustly express both Flk1 and cKit, which are established early markers of stem-cell-ness (5,20,31,41). In addition, wt and GRMD CPCs express NG2, αSMA, PDGFRα, and PDGFRβ, suggesting pericyte origin. Based on our results, Ven CPCs recapitulate both mesoangioblast and cardiac stem cell markers. The absence of pluripotency genes expression and surface markers such as CD73 and CD105 excludes our cells from definition as cardiac stromal cells (CStCs) (43). The analysis of mesodermal differentiation potential reveals that those cells that were able to differentiate into smooth muscle cells, adipocytes, and endothelial cells failed to generate osteoblasts also when stimulated with BMPs. The endothelial differentiation ability has already been observed in murine cardiac stem cells (20), but in canine cells the phenomenon is more similar to their human counterpart (19). As expected, all clones promptly responded to TGF-β by activating a smooth muscle differentiation program. The differentiation of Ven CPCs into myocytes has been tested with different approaches, including cocultures and genetic manipulation. As expected, only wt but not GRMD Ven CPCs can express dystrophin at differentiation endpoints. Differences in cardiomyogenic potential were observed between wt and GRMD Ven CPCs when using serum starvation, azacytidine treatment, and expression of Nkx2.5. Nevertheless, all the protocols tested failed to induce ion channel expression levels and the formation of beating cardiomyocytes. No relevant differences were observed among wt and GRMD Ven CPCs in terms of in vitro cardiomyocyte proliferation or growth factor secretion, and thus, we speculated that these cells would have an equal contribution in terms of paracrine effects. Previous studies featuring resident cardiac stem cells that are capable of repairing the infarcted heart have been viewed with skepticism and still some questions remain. In fact, the poor capability of cardiac regeneration during acute ischemic events, in which the scar tissue formation is predominant, places doubt on the value of resident stem cells in the myocardium. Thus, evaluating the differentiation potential of CPCs in an animal model of chronic cardiomyopathy is necessary to understand their functional properties. In this study, we provide evidence that the adult canine heart contains a growth reserve that is represented by CPCs. These cells may eventually be coaxed to home to the necrotic area and participate to cardiac regeneration, although this is limited to the surviving portion surrounding the infarcted area (47). In the last few years, several stem cell types have been challenged to rescue the phenotype of preclinical models of acute heart damage and resident CPCs have been identified in the heart of mouse, hamster, rat, pig, dog, and humans. In this regard, our study advances the field of regenerative cardiology, showing for the first time that CPCs can be isolated from a large animal model of cardiomyopathy. Moreover, the demonstration that cardiac microRNAs (miR-1, miR-133a and miR-208) are downregulated in GRMD CPCs identifies possible therapeutic targets for gene therapy. miR-21 upregulation in GRMD CPCs is also intriguing, since it has recently been shown to be a key element in the fibrotic program of several tissues, including lung (32) and heart (49). It has also been reported that miR-21 positively regulates TGF-β signaling by directly targeting SMAD7 (mothers against decapentaplegic homolog 7), explaining at least in part the molecular mechanism of miR-21-mediated fibrosis (34). We identified an upregulation of miR-21 in GRMD Ven CPCs that is consistent with the literature, and consequently, miR-21 could be a potential target to correct undesired fibrotic differentiation events. Additionally, the overexpression of miR-21 in GRMD CPCs raises interesting questions if the scar tissue of an infarcted heart is generated exclusively by the proliferation of connective tissue fibroblasts. We could speculate that the exhaustion of CPC cardiomyogenic potential, due to several attempts to replace damaged cardiomyocytes, leads to a shortened lifespan and an altered differentiation program that is also observed in vitro after several doublings. For these reasons, GRMD CPCs may be responsible, at least in part, for scar tissue formation in the cardiomyopathic heart. Nevertheless, cardiac integration of both wt and GRMD CPCs observed in newborn SCID/beige transplanted mice fulfilled their cardiac differentiation potential. As opposed to newborn mice treated with canine fibroblasts, CPCs-treated mice showed normal cardiac architecture compared with sham-operated mice. However, GRMD CPCs failed to functionally integrate in the murine myocardium at comparable levels with wt progenitors, confirming the differentiation impairment observed in vitro. In fact, many dsRed+ nuclei of wt Ven CPCs tend to localize in the dystrophin-positive area of the host myocardium whereas dsRed+ nuclei coming from GRMD Ven CPCs seem to be outside the dystrophin-positive cells. GRMD CPCs transplantation increases the percentage of donor-derived interstitial cells, presumably due to the activation of an altered fibrogenic program (i.e., overexpression of miR-21). In our opinion, this represents a critical aspect to be considered for a tailored cell therapy using autologous approaches (23,48,51). An ideal candidate cell for the treatment of muscular dystrophy-related cardiomyopathies is one that has restored expression of a missing gene, such as dystrophin, in this case. In fact, we documented dystrophin expression by wt Ven CPCs both in vitro and in vivo. Our xenograft in vivo study is relevant for two reasons. The first is the demonstration that 6-month-old CPCs still have cardiomyogenic potential in vivo, and second is the proof of concept that encourages a move towards cell based transplantation protocols that target the GRMD heart. This is a critical issue considering that a clinical trial for muscular dystrophy is currently running after the promising results were obtained after treating GRMD skeletal muscles with mesoangioblasts (45,46).
Until now, the use of circulating progenitors as therapeutic tools has provided good support for paracrine-mediated benefits. The discovery of local cardiac committed stem cells opens a new scenario in terms of cardiac tissue replacement. As these cells tend to disappear from the heart of older (19) or cardiomyopathic mice (14), a therapeutic approach using CPCs should be directed as early as possible to target the necrotic area (59). In fact, for such a diffusely damaged cardiac area, as observed in GRMD dogs or in large transmural infarctions, CPCs may not be able to heal the whole area, and neoangiogenesis and removal of necrotic tissues may be required to allow for correct cell migration. CPC isolation methods have already been described for mouse (20), human (19), and now for the dystrophic dog and can be applied extensively for the human heart to generate more data regarding committed human cardiac progenitors, moving the field towards tailored cell therapy. This is particularly relevant for progressive pediatric cardiac diseases and to avoid the risks inherent to the systemic mobilization of circulating stem cells, such as homing to nontarget organs.
In conclusion, cellular therapies need to become organ-specific, and here we accomplished at least three objectives that move the cardiac stem cell field forward. Firstly, we provide evidence that cardiac progenitors can be isolated from a large animal model of cardiac degeneration, such as the dystrophic GRMD heart. Nonetheless, the use of a stably integrating nonviral gene marking system based on transposons provides a technological improvement that facilitates in vivo tracing studies. Finally, the data from the present study provides new insights towards the application of cardiac CPCs for cell therapy of progressive cardiac diseases.
Footnotes
Acknowledgments
We thank Gianpaolo Papaccio and Mark Guns for critical discussion and Shea Carter for the editing of the manuscript. We thank Ermira Samara-Kuko and Rudi Micheletti for the technical assistance, Christina Vochten for the professional secretarial service, and Paolo Luban for a kind donation. This work was supported by the Nash Avery Stem Cell Research–Wicka Fund, University of Minnesota; the Fonds Wetenschappelijk Onderzoek Odysseus Program grant G.0907.08; Research Council of the University of Leuven grant OT/09/053; Cardio Repair European Multidisciplinary Initiative grant 242038 FP7-EC; the Italian Ministry of University and Scientific Research grant 2008RFNT8T_003 (Progetto di Ricerca di Interesse Nazionale 2008); and EC (CARE-MI) FP7-HEALTH-2011.1.4-2-two-stage and Cariplo grants 2007.5639 and 2008.2005 to M. Sampaolesi. The authors declare no potential conflict of interest.