
Abstract
Amyotrophic lateral sclerosis (ALS) is characterized by progressive dysfunction and degeneration of motor neurons in the central nervous system (CNS). In the absence of effective drug treatments for ALS, stem cell treatment has emerged as a candidate therapy for this disease. To date, however, there is no consensus protocol that stipulates stem cell types, transplantation timing, or frequency. Using an ALS mouse model carrying a high copy number of a mutant human superoxide dismutase-1 (SOD1)G93A transgene, we investigated the effect of neural induction on the innate therapeutic potential of mesenchymal stem cells (MSCs) in relation to preclinical transplantation parameters. In our study, the expression of monocyte chemoattractant protein-1 (MCP-1) was elevated in the ALS mouse spinal cord. Neural induction of MSCs with neurogenin 1 (Ngn1) upregulated the expression level of the MCP-1 receptor, CCR2, and enhanced the migration activity toward MCP-1 in vitro. Ngn1-expressing MSCs (MSCs-Ngn1) showed a corresponding increase in tropism to the CNS after systemic transplantation in ALS mice. Notably, MSCs-Ngn1 delayed disease onset if transplanted during preonset ages, whereas unprocessed MSCs failed to do so. If transplanted near the onset ages, a single treatment with MSCs-Ngn1 was sufficient to enhance motor functions during the symptomatic period (15–17 weeks), whereas unprocessed MSCs required repeated transplantation to achieve similar levels of motor function improvement. Our data indicate that systemically transplanted MSCs-Ngn1 can migrate to the CNS and exert beneficial effects on host neural cells for an extended period of time through paracrine functions, suggesting a potential benefit of neural induction of transplanted MSCs in long-term treatment of ALS.
Keywords
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset degenerative disease that is characterized by the death of motor neurons in the cortex, brain stem, and spinal cord. Patients with ALS exhibit progressive muscle atrophy and respiratory paralysis and usually die within 3–5 years after the first appearance of symptoms (8,9,53,55). About 10% of ALS patients are diagnosed with inherited familial ALS (fALS), and 20% of fALS cases are associated with mutations in the Cu2+/Zn2+ superoxide dismutase (SOD1) gene (54). The remaining 90% of patients are diagnosed with sporadic ALS, which is associated with diverse factors, including oxidative stress and glutamate excitotoxicity (57), viral infection (7), autoimmune mechanisms (3), abnormal accumulation of neurofilaments (63), altered glial function (11,44), mitochondrial dysfunction (42), and impaired trophic support of the host environment (25). The diverse etiology of ALS makes effective treatment difficult.
Recently, stem cells have emerged as therapeutic candidates for the treatment of neurological diseases. Mesenchymal stem cells (MSCs) from bone marrow are known to be effective in improving neurological functions in stroke patients (5). MSC transplantation has also yielded positive results in the SOD1 mutant mouse model of ALS (60,72) and is potentially a viable and safe therapeutic strategy for patients with ALS (17,40).
Although early phase clinical trials utilizing MSCs as therapeutic cell sources for the treatment of ALS patients are ongoing, there are no consensus protocols for stem cell therapy that stipulate what kinds of stem cells should be used, when transplantation of stem cells should be performed, or how many cells should be transplanted. Human neuron-committed teratocarcinoma (hNT) cells (19–21,66) and neural stem cells (13,38,45,68) have been reported to delay disease progression more effectively than MSCs of nonneuronal origin in ALS animal models, suggesting the superiority of neural lineage cells compared to mesodermal lineage cells in treating ALS.
Whether MSCs can be induced to differentiate into ectodermal neuronal lineages by treatment with chemicals and growth factors is a matter of controversy (14,35,46, 50,67). Previously, we reported that neurogenin 1 (Ngn1), a proneural basic helix–loop–helix transcription factor, was sufficient to reprogram the mesodermal cell fate of human MSCs into a neural fate (30). Ngn1-induced MSCs to express markers of neuronal phenotypes, such as NeuroD and voltage-gated Ca2+ and Na+ channels. Moreover, in an animal stroke model, Ngn1 enhanced the therapeutic effects of MSCs by directing the cells to differentiate into neurons and integrate into the host neural circuit. Ngn1 did so without altering the innate paracrine functions of MSCs, such as protecting cells, promoting neurogenesis, and suppressing immune responses (30). Taken together, these observations suggest that overexpression of Ngn1 may provide a reliable means to endow MSCs with neural properties and thereby enhance their therapeutic potential against ALS.
In the present study, we used a hSOD1G93A transgenic mouse model (23) to compare the effectiveness of non-neural (unprocessed) MSCs and neurally induced, Ngn1-expressing MSCs (MSCs-Ngn1) in treating ALS. We also characterized critical parameters that are likely to affect the clinical effectiveness of stem cell therapy, including transplantation timing and frequency.
Materials and Methods
MSCs-Ngn1 and In Vitro Differentiation
Human MSCs were isolated from bone marrow obtained from the iliac crest of six healthy 10- to 15-year-old male donors with informed consent and approval of the Institutional Review Board of Ajou University Medical Center, by plastic adherence, as described previously (29). Cells were cultured in growth medium [Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 10 ng/ml basic fibroblast growth factor (bFGF; Dong-A Pharmaceutical Co., Youngin, Korea)]. MSCs-Ngn1 were prepared by transducing MSCs with a retroviral vector encoding Ngn1, as previously described (30), and were maintained in growth medium.
Neural induction by Ngn1 was verified by monitoring the terminal differentiation of MSCs-Ngn1 into neuronal cells in vitro. Terminal differentiation was induced by growing MSCs-Ngn1 on poly-d-lysine/collagen-coated coverslips in growth medium containing 10 μM 5-azadeoxycytidine (Aza-dC; Sigma-Aldrich, St. Louis, MO, USA) for 3 days and then in DMEM/F12 (Invitrogen) containing 10% FBS and 10 μM forskolin (Sigma-Aldrich) for 14 days. Neuronal differentiation was verified by immunocytochemistry using antibodies against the neuronal markers microtubule-associated protein 2 (MAP2; 1:500, Sigma-Aldrich), neurofilament 200 (NF200; 1:500, Sigma-Aldrich), and neuronal nuclei (NeuN; 1:100, Chemicon, Temecula, CA, USA).
ALS Animals
Transgenic mice harboring a high copy number of the hSOD1G93A [B6SJL-TgN(SOD1-G93A)1Gur] transgene, described by Gurney et al. (23), were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Hemizygous transgenic progeny were maintained by mating transgenic males with F1 hybrid females, obtained by crossing C57BL6 females with Swiss Jim Lambert (SJL) males. Genotypes were verified by polymerase chain reaction (PCR) using genomic DNA isolated from mouse tail extracts (12). Amplifications (35 cycles; annealing temperature, 60°C) were performed using forward (5′-CAT CAG CCC TAA TCC ATC TGA-3′) and reverse (5′-CGC GAC TAA CAA TCA AAG TGA-3′) primers specific for the hSOD1G93A transgene. Food and water were provided ad libitum, and all experimental procedures were reviewed and approved by the Institutional Animal Research Ethics Committee at the Ajou University Medical Center (Suwon, South Korea).
Transplantation
In each experiment, mice were divided into three groups matched by age and gender. Mice received lateral tail vein injections of 1 × 106 unprocessed MSCs or MSCs-Ngn1 in 0.1 ml phosphate-buffered saline (PBS). Experiments were carried out using three transplantation protocols (Table 1). In the first set of experiments (Experiment I), animals received cells before the onset of ALS symptoms (8 weeks after birth). In the second experiment (Experiment II), cells were injected during the same week that animals first failed the paw grip endurance (PaGE) test (14th–16th weeks). In the third experiment (Experiment III), animals received cells twice—once at 13 weeks and once at 15 weeks. The animals in this experiment were also intraperitoneally (IP) injected with 4 mg/kg cyclosporine A (CsA; Chong Kun Dang Pharm., Seoul, Korea) immediately prior to first transplantation and every other day thereafter. The negative control group received PBS instead of cells.
Experimental Protocols and Summary Results

Data are presented as means ± SEMs. (*p<0.05, **p<0.01; Tukey's HSD). PBS, phosphate buffered saline (control); MSCs-Ngnl, neurogenin-1 expressing mesenchymal stem cells; M, male; F, female; CsA, cyclosporine A; PaGE; paw grip endurance test.
Identification of transplanted cells
For the identification of transplanted cells in the mouse, we transduced cells with adenoviral vectors expressing nuclear localization signal (NLS)-linked β-galactosidase (LacZ; multiplicity of infection = 100) which was kindly provided by Dr. YH Kim (Kyung Hee University, Seoul, Korea), prior to transplantation. Animals transplanted at 8 weeks, as described above, were sacrificed 2 weeks later by transcardial perfusion with 0.9% saline and 2% paraformaldehyde (Invitrogen) in 0.1 M phosphate buffer (pH 7.4) under deep anesthesia. The brain, spinal cord, liver, heart, lung, kidney, spleen, and muscle tissues were trimmed to a thickness of 3–4 mm, postfixed in 2% paraformaldehyde for 12 h, and incubated at 37°C overnight in X-gal staining solution [KOMA Biotech, Seoul, Korea; 1 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-d-galactopyranoside), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 2 mM MgCl2, 0.02% Nonidet P-40, 0.1 M phosphate buffer, pH 7.4]. Tissues were embedded in paraffin and serially sectioned into 5-μm sections for histological analysis.
RNA Isolation and RT-PCR
Total RNA was extracted from homogenized frozen spinal cord specimens or cells with RNAzol B (Tel-Test; Friendswood, TX, USA), and cDNA was synthesized from 1 μg total RNA using the First Strand cDNA Synthesis Kit (Roche Diagnostics, Mannhein, Germany), according to the manufacturer's recommendations. PCR amplifications were subsequently performed using specific primers (summarized in Table 2).
Primers and PCR Protocol
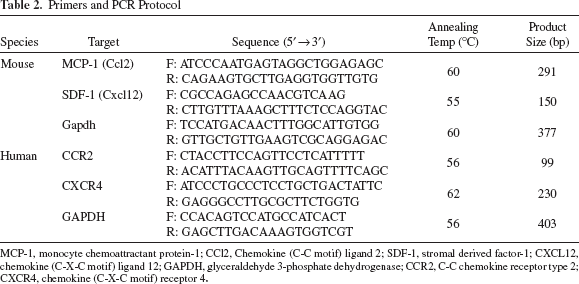
MCP-1, monocyte chemoattractant protein-1; CCl2, Chemokine (C-C motif) ligand 2; SDF-1, stromal derived factor-1; CXCL12, chemokine (C-X-C motif) ligand 12; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; CCR2, C-C chemokine receptor type 2; CXCR4, chemokine (C-X-C motif) receptor 4.
Cell Migration Assays
Migration assays were performed in Transwell cell culture chambers with polycarbonate filters (six-well, 8-μm pore; Corning Costar, Cambridge, MA, USA). The upper side of the Transwell filter was coated with 30% growth factor-reduced Matrigel (#354230; BD Bioscience, San Jose, CA, USA) in PBS for 30 min at 37°C. Cells were placed in the upper compartment of the chamber at a density of 2×105 cells in 1 ml of assay media (DMEM with 2% FBS), and monocyte chemoattractant protein-1 (100 ng/ml MCP-1/JE; R&D Systems, Minneapolis, MN, USA) was placed in the lower compartment. To verify the specificity of MCP-1, a neutralizing anti-MCP-1 antibody (20 μg/ml; R&D Systems) was added to the lower chamber. After incubating for 24 h at 37°C in a humidified 5% CO2 environment, the cells on the filters were fixed and stained with a 0.5% crystal violet solution (Sigma-Aldrich). Nonmigrated cells on the upper surface of the filter were removed with a cotton swab, and the remaining cells on the lower surface were counted in 10 random fields under a light microscope (100x).
Quantification of Motor Neurons and Transplanted Cells
The neuroprotective effects of unprocessed MSCs and MSCs-Ngn1 were assessed by semiquantitative evaluation of motor neurons and transplanted cells in the spinal cord. The animals were transplanted with cells at 8 weeks of age and were sacrificed at 16 weeks. The cervical and lumbar segments of the spinal cord were isolated and postfixed with 2% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) and cryoprotected in 30% sucrose in 0.1 M phosphate buffer (pH 7.4) overnight. Ten 30-μm-thick sections at 300-μm intervals were obtained per cervical or lumbar spinal cord from each animal. For quantifying the motor neuron survival, sections were stained sections stained for Nissl using cresyl violet (Sigma-Aldrich). All large cells (diameter > 20 μm) containing a distinct nucleus, prominent nucleoli, and at least onet thick process in the ventral horn below a lateral line from the central canal were counted. To identify ventral motor neuron, adjacent sections were immunostained with anti-choline acetyltransferase antibody (ChAT; 1:200, Chemicon), and antibody reactions were visualized using an ABC kit (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer's instructions.
For counting the number of grafted cells, sections were immunostained with a primary antibody against human mitochondria (hMT; Chemicon; 1:100). After washing, immunoreactivity was visualized using an ABC kit (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer's instruction. The number of immunoreactive cells was counted in five sections each of the cervical and lumbar region of spinal cord. Data are presented as means ± standard errors of the mean (SEMs) from five mice per group.
Characterization of grafted cells was performed by double-immunofluorescence staining using cell type-specific antibodies. Briefly, sections were blocked with 10% normal serum in PBS and incubated with antibodies against NF200 (1:200, Sigma-Aldrich), glial fibrillary acidic protein (GFAP; 1:200, Sigma-Aldrich), ionized calcium binding adaptor molecule 1 (Iba1; 1:100, Wako Pure Chemical, Chou-Ku, Osaka, Japan), and/or hMT (1:100, Chemicon) at 4°C overnight. Sections were then incubated with Alexa Fluor 488- or 594-conjugated anti-IgG secondary antibodies (1:2,000, Molecular Probes, Eugene, OR, USA) and counterstained with bisbenzamide (Hoechst; Molecular Probes) to visualize the entire population of cells. Fluorescent images were acquired using a Zeiss LSM710 confocal microscope (Carl Zeiss, Jena, Germany).
Evaluation of Motor Functions
In all experiments, independent investigators were double-blinded to the animal's pre- and posttransplant status to avoid subjective bias.
Extension reflexes were evaluated according to the procedure and scoring system described by Barneoud et al. (6) and Weydt et al. (64). Briefly, mice were suspended by the tail, and the degree of motor deficit was scored from 0 to 2 as follows: normal extension reflex in both hindlimbs, 2; imbalanced extension in the hindlimbs, 1.5; extension reflex in only one hindlimb, 1.0; absence of any hindlimb extension, 0.5; and total paralysis, 0.
The basic motor functions, balance and grip strength, were measured using the PaGE test (64), in which the latency to falling is measured for a mouse holding onto the inverted lid of a cage. Each mouse was given three trials, and the longest latency was recorded. The cutoff time was 90 s.
Performance of a complex task involving motor coordination, balance, and strength (6,43) was assessed using a rotarod apparatus (Acceler Rota-Rod 7650, UGO BASILE, Varese, Italy) with a 3.5-cm-diameter rod revolving at a constant speed of 16 rpm. Each animal was given three trials, and the longest latency before falling was recorded. An arbitrary cutoff time was set at 300 s.
Disease-onset time was defined as the time at which the mouse could no longer remain on the rotarod apparatus for 300 s at a speed of 16 rpm. Mortality was scored as the age of death or the age at which the mouse was unable to right itself within 30 s after being placed on its side.
Statistical Analysis
Statistical analyses were carried out using SPSS version 15.0 (SPSS, Inc., Chicago, IL, USA). Data were analyzed by Student's t test or one-way analysis of variance (ANOVA), as appropriate. Significant differences were further evaluated using Tukey's HSD post hoc test. All results are expressed as means ± SEMs.
Results
In Vitro Cultivation and Neural Induction of MSCs
Human bone marrow MSCs were neurally induced by stably transducing with a retroviral vector encoding Ngn1, as described in Materials and Methods. Consistent with our previous results (30), the resulting MSCs-Ngn1 exhibited a neuron-like morphology, exemplified by the expression of the neuron-specific proteins NeuN, NF200, and MAP2 (Fig. 1). Under the same conditions, these proteins were not detected in unprocessed MSCs.

Neural induction of MSCs with Ngn1. Cells were induced to differentiate into neuronal cells as described in Materials and Methods. Immunocytochemistry revealed expression of neurofilament 200 (NF200), microtubule associated protein 2 (MAP2), and Neuronal Nuclei (NeuN) (green) in neurogenin-1 transduced mesenchymal stem cells (MSCs-Ngn1) but not nontransduced MSCs. Nuclei were counter stained with Hoechst (blue). Scale bar: 50 μm.
Distribution of Transplanted MSCs-Ngn1 in the Central Nervous System
Motor neurons begin to degenerate in a diffuse manner over the entire central nervous system (CNS) of hSOD1G93A mice starting at 9 weeks of age (38). MSCs-Ngn1 or unprocessed MSCs were systemically delivered to the CNS through tail–vein injection of 1 × 1 0 6 cells at 8 weeks of age, a time at which the animals had not yet exhibited ALS symptom. The cells were prelabeled with adenoviral vectors encoding an NLS-linked LacZ gene for identification of grafted cells. Two weeks after intravenous administration, the mice were sacrificed and LacZ gene expression in tissues was assessed by X-gal staining (Fig. 2). X-gal-positive MSCs-Ngn1 were often found in the liver, but not in the spleen, lung, kidney, heart, or muscle (Fig. 2C). Cells positive for X-gal were readily detected along the entire CNS of animals injected with MSCs-Ngn1 (Fig. 2A, B), but to a much lesser extent in animals injected with unprocessed MSCs (data not shown).

Tissue distribution of grafted MSCs-Ngn1. MSCs-Ngn1 were prelabeled with LacZ and systemically injected into the tail vein at 8 weeks. After 2 weeks, (A, B) β-Galactosidase (X-gal) staining revealed the presence of grafted cells in the striatum, midbrain, brainstem, and spinal cord. (C) MSCs-Ngn1 were also found in the liver, but not in the kidney, heart, skeletal muscle, spleen, or lung. Scale bar: 20 μm.
Analysis of the Migration Mechanism
To better understand the targeting mechanisms of grafted cells, we first investigated the expression of monocyte chemotactic protein-1 (MCP-1) and stromal derived factor-1 (SDF-1), which are known to be important chemoattractants for MSCs migration (58,61). Total RNA was extracted from homogenized spinal cord tissues of wild-type (WT) and hSOD1G93A mice at ALS onset ages. RT-PCR analyses revealed that MCP-1 expression was enhanced in hSOD1G93A mice compared to WT littermates. In contrast, SDF-1 was constitutively expressed regardless of the presence of the hSOD1G93A gene (Fig. 3A). Next, we examined the expression of chemokine receptors by MSCs and MSCs-Ngn1, focusing on CCR2 (CC-motif) and CXCR4 (CXC-motif) chemokine receptors for MCP-1 and SDF-1. CCR2 and CXCR4 were expressed in MSCs and were present at even higher levels in MSCs-Ngn1 (Fig. 3B). These data suggest that the greater tropism of MSCs-Ngn1 to the CNS of ALS mice can be partly ascribed to the enhanced interaction of MCP-1 and CCR2. To further verify that increased expression of CCR2 could contribute to the enhanced migratory activity of MSCs-Ngn1, we carried out in vitro (Transwell) migration assays. The cells were seeded in the upper Transwell chamber, while recombinant MCP-1 was added to the lower chamber (Fig. 3C). MSCs exhibited little migration to the lower chamber in the absence of MCP-1 but showed substantial migratory activity in the presence of MCP-1. By comparison, the migratory potential of MSCs-Ngn1 was high in the absence of MCP-1 and was further increased by MCP-1. Addition of a neutralizing anti-MCP-1 antibody interfered with the migration of MSCs-Ngn1 to the lower chamber, specifically implicating MCP-1 in the observed chemoattraction. These results suggest the possibility that the MCP-1/CCR2 axis plays an essential role in enhancing the migration of MSCs-Ngn1 to the ALS spinal cord. It should be emphasized that both spontaneous and chemotactic migration were significantly enhanced by neural induction of MSCs with Ngn1 compared with that in unprocessed MSCs.

Detailed analysis of the migration mechanism. (A) RT-PCR analysis of mRNA transcripts for monocyte chemoattractant protein-1 (MCP-1) and stromal derived factor-1 (SDF-1) in the lumbar spinal cord of human G93A mutated superoxide dismutase (hSOD1G93A) and wild-type (WT) mice at 13 weeks. Note that the MCP-1 expression was dramatically increased in presymptomatic amyotrophic lateral sclerosis (ALS) mice, whereas the SDF-1 expression was not different between hSOD1G93A mice and WT. Distilled water (dH2O) was used as a negative control. (B) RT-PCR analyses indicated that unprocessed MSCs and MSCs-Ngn1 express cytokine receptors, including C-C chemokine receptor type 2 (CCR2) and chemokine (C-X-C motif) receptor 4 (CXCR4). dH2O was used as a negative control. (C) Transwell migration assays were used to assess the migratory activity of MSCs and MSCs-Ngn1 toward MCP-1. Both spontaneous and chemotactic migration were enhanced in MSCs-Ngn1. Data are presented as means ± SEMs (**p < 0.01; Student's t test).
Effects of Transplanted Cells on Spinal Motor Neuron Degeneration
To assess the protective effects of unprocessed MSCs and MSCs-Ngn1 before the onset of ALS symptoms, we compared the survival rates of spinal motor neurons by Nissl staining at 16 weeks of age in animals that had received 1 × 106 cells at 8 weeks in the tail vein (Fig. 4A, B). Cells in the ventral horn with a cell body greater than 20 μm in diameter and a distinct nucleus also expressed ChAT (data not shown), indicating that they were ventral motor neurons. In PBS-injected hSOD1G93A control mice, the number of these motor neurons in the cervical and lumbar regions was reduced to 25% and 15%, respectively, of the numbers in healthy WT littermates (WT cervical: 58 ± 2.1, PBS cervical: 14 ± 1.0; WT lumbar: 51 ± 2.2, PBS lumbar: 8 ± 1.2). Notably, transplantation of MSCs-Ngn1 prevented the loss of ventral motor neurons and increased cervical and lumbar neuron survival to 38% and 24%, respectively, of that in WT littermates (cervical: 22 ± 1.1, lumbar: 12 ± 1.1). In contrast, transplantation of unprocessed MSCs did not prevent motor neuron loss (cervical: 13 ± 0.7, lumbar: 7 ± 0.7).

Protection of motor neurons by MSCs-Ngn1. Animals received cells or PBS through tail vein injections at 8 weeks and sacrificed at 16 weeks for the analysis of spinal cord. (A) Representative microphotograph of Nissl staining or (C) human mitochondria (hMT) immunohistochemical staining in the ventral horn of cervical and lumbar spinal cords of hSOD1G93A mice receiving unprocessed MSCs or MSCs-Ngn1. Arrowheads denote donor-derived hMT positive cells. Quantitative cell counts of motor neurons and transplanted cells in the spinal cord are displayed in diagram (B) and (D), respectively. Data are presented as means ± SEMs from at least five animals. (**p < 0.01; Tukey's HSD). (E–G) Confocal images of grafted cells in the cervical spinal cords of hSOD1G93A mice treated MSCs-Ngn1. None of the MSCs-Ngn1 that were identified with anti-hMT antibody (green) costained with antibodies recognizing NF200 (E), glial fibrillary acidic protein (GFAP)(F), or ionized calcium binding adaptor molecule 1 (Iba1)(G; red). Scale bar: 100 μm.
To compare the integration efficiency of unprocessed MSCs and MSCs-Ngn1 into the spinal cord of hSOD1G93A mice, we counted hMT immunoreactive cells in the cervical and lumbar sections of spinal cord. The immune reactive cells were readily detected in the spinal cord parenchyma of animals injected with MSCs-Ngn1 but to a lesser extent in animals injected with unprocessed MSCs (Fig. 4C, D). Surprisingly, most grafted cells in the spinal cord did not express GFAP (an astrocyte marker), NF200 (a neuronal marker), or Iba1 (a microglia and macrophage marker) (Fig. 4E–G). The results suggest that transplanted MSCs-Ngn1 had the potential to efficiently migrate from the circulation into CNS regions, where they persist to remain as undifferentiated cells.
Delayed Disease Progression by Treatment with MSCs-Ngn1
To determine if histological improvement correlated with behavioral improvement, we evaluated behavior using three different experimental frameworks (Table 1). For the Experiment I, we randomized the animals and transplanted the cells or PBS to each group. We systemically injected cells into the tail vein at a preonset stage (8 weeks) and monitored animals until death (up to 18–20 weeks) (Fig. 5A). Early transplantation of MSCs-Ngn1 (i.e., before motor neurons started to degenerate) delayed the average age of disease onset by 5 days (16.8 vs. 16.1 weeks) and extended the average life span by 3 days (18.6 vs. 18.2 weeks) (Fig. 5B, C). Importantly, the group treated with MSCs-Ngn1 performed better on extension reflex, rotarod, and PaGE tests during the symptomatic period (15–17 weeks) than did the PBS-treated control group (Fig. 5D–F) (Table 1, Experiment I). In contrast, unpro cessed MSCs did not alter disease-onset age or life span but did improve extension reflex test scores (Fig. 5D). This result is similar to the increased survival of ventral motor neu-rons in animals administered MSCs-Ngn1 compared to those injected with unprocessed MSCs (Fig. 4). These results indicate that early transplantation of MSCs-Ngn1 may effectively delay disease onset and moderately improve motor functions in xenograft animal models.

The effects of transplantation of unprocessed MSCs and MSCs-Ngn1 at preonset ages. (A) Cells were systemically injected into the tail vein at 8 weeks (T.P) and mice were monitored from disease onset to endpoint stages (18–20 weeks). MSCs-Ngn1 delayed disease onset (defined as the date of first failed rotarod test) (B) and extended life span by 3 days (C). MSCs-Ngn1 moderately increased motor performance in extension reflex (D), rotarod (E), and paw grip endurance (PaGE)(F) tests, whereas unprocessed MSCs increased scores only in the extension reflex test (D). Data are presented as means ± SEMs from eight animals.
Next, we investigated the influence of transplantation timing on disease progression. In this experiment (Table 1, Experiment II) (Fig. 6A), we randomly grouped hSOD1G93A mice based on their performance in PaGE test, which appeared to be a predictive indicator of the disease onset. Thus, we were able to prepare and transplant cells near the onset ages. Transplantation of MSCs-Ngn1 increased the average life span by 7 days (from 18.2 to 19.3 weeks) (Fig. 6B). Moreover, the MSCs-Ngn1-treated group showed outstanding performance in all tests throughout the symptomatic period (15–17 weeks) (Fig. 6C–E, filled bars). Although the group treated with unprocessed MSCs showed moderately higher performance in rotarod test than did the PBS-treated group at 16 weeks (unprocessed MSCs: 203.6 ± 35.9 s; PBS: 116.0 ± 45.3 s), this difference was not maintained through 17 weeks. Collectively, these results indicate that neural induction with Ngn1 extended the therapeutic effects of MSCs with respect to the delay of disease progression. The results also suggest that treatment timing is important for cell therapy of ALS: the therapeutic effects were more prolonged when MSCs-Ngn1 were transplanted at onset rather than preonset ages (see Discussion).

Comparison of the therapeutic potential of unprocessed MSCs and MSCs-Ngn1 transplanted near the onset ages. (A) Cells were systemically injected into the tail vein immediately after the first failure in the PaGE test (14th–16th weeks). Transplantation of MSCs-Ngn1 not only extended life span (B) but also enhanced motor performance during symptomatic ages (15–17 weeks), as determined by extension reflex (C), rotarod (D), and PaGE tests (E). In contrast, unprocessed MSCs moderately increased motor performance only at 16 weeks. Data are presented as means ± SEMs from eight animals (**p < 0.01; Tukey's HSD).
Enhanced Therapeutic Effects of Repeated Grafting of MSCs-Ngn1
To test whether the short-lived effects of MSCs could be extended by multiple treatments, we randomized the animals into three groups and transplanted cells twice, first at 13 weeks of age and then at 15 weeks (Fig. 7). In Experiment III, all animals were immunosuppressed with CsA (4 mg/kg IP every other day) to eliminate immune rejection that might be evoked by repeated grafting (69). Consistent with previous reports (24), treatment with CsA alone did not influence disease progression; survival rates and behavioral scores did not differ between CsA- and vehicle-treated groups (Fig. 8). When treated twice, the therapeutic effects of MSCs-Ngn1 were more prominent. MSCs-Ngn1 extended the average life span by more than 9 days compared with the PBS control (19.1 vs. 17.8 weeks). The scores for extension reflex, rotarod, and PaGE tests were significantly higher in animals that received two injections of MSCs-Ngn1 between 15 and 16 weeks than in PBS-treated controls (p < 0.01). Importantly, when injected twice, unprocessed MSCs were as effective at 15–16 weeks in improving PaGE, extension reflex, and rotarod test scores as were MSCs-Ngn1. Our results suggest that the first treatment of MSCs-Ngn1 at 13 weeks delayed disease onset, and the second treatment ensured improvement in behavioral performance during the symptomatic period.
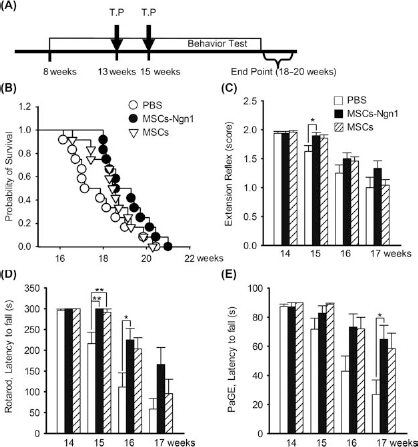
Effects of multiple MSC transplantations. (A) Cells were systemically injected twice into the tail vein—once at 13 weeks and once at 15 weeks. Transplantation of MSCs-Ngn1 not only increased survival rates (B) but also enhanced motor functions at a higher level of statistical confidence during symptomatic ages (15–17 weeks), as determined by extension reflex (C), rotarod (D), and PaGE tests (E). Notably, the scores for the rotarod test were significantly higher in animals that received two injections of unprocessed MSCs at 15 weeks than in PBS-treated controls (D). Data are presented as means ± SEMs from 12 animals (*p < 0.05, **p < 0.01; Tukey's HSD).
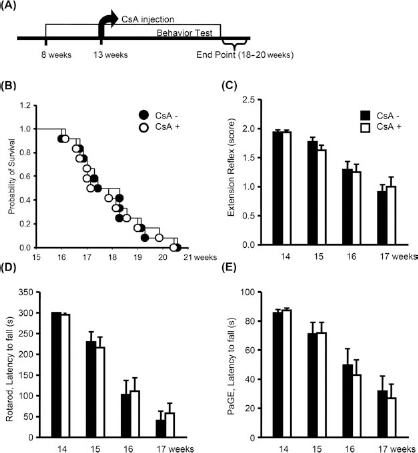
Effects of CsA on disease progression. Animals were injected (IP) with 4 mg/kg cyclosporine A (CsA) every 2 days starting from 8 weeks of ages and continuing until the endpoint (A). CsA alone did not extend life span (B) or motor performance, as measured by extension reflex (C), rotarod (D), and PaGE tests (E). Data are presented as means ± SEMs from 12 animals.
Discussion
The present study demonstrates that transplantation of MSCs into the systemic circulation can delay motor impairment in an ALS animal model for a limited period of time and that neural induction with Ngn1 can extend the short-lived beneficial effects of MSCs and efficiently delay disease progression.
MSCs have recently emerged as a therapeutic candidate for the treatment of a variety of disorders, including myocardial infarction (31,39), skeletal muscle injury (49), brain injury (37,73), neurodegenerative diseases (33), spinal cord injury (51), and peripheral nerve injury (59). The therapeutic benefits of MSCs in pathological conditions are mostly mediated through the release of neurotrophic factors, including brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor (GDNF), ciliary neurotrophic factor (CNTF), bFGF, vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), and insulin-like growth factor (IGF) (32,47,65).
We previously showed that treatment with MSCs improved recovery in an animal stroke model by exerting paracrine functions. Intracranial transplantation of MSCs reduced neuronal cell death in the host brain, promoted endogenous neurogenesis, and suppressed inflammatory responses (70). In the same model, we also showed that the therapeutic effects of MSCs were enhanced by neural induction of cells with Ngn1 (30). MSCs-Ngn1 transdifferentiated into neurons and integrated into host neural circuits while maintaining the original paracrine functions of unprocessed MSCs. In contrast to the stroke model, we could not find neuronal cells derived from MSCs-Ngn1 in hSOD1G93A mice (Fig. 4E, F), suggesting that MSCs-Ngn1 react differentially to the distinctive pathological host microenvironments associated with progressive diseases (e.g., ALS) and acute brain injury (e.g., stroke) (52). Nevertheless, MSCs-Ngn1 could integrate into the CNS of ALS animals with higher efficiency than unprocessed MSCs (Fig. 4C, D), and showed greater protection of spinal motor neurons (Fig. 4A, B). The higher survival of motor neurons in the animals with MSCs-Ngn1 correlates well with the preserved motor functions at 16 weeks (Fig. 5D, E). Consistent with these data, when transplanted around onset ages, MSCs-Ngn1 delayed the first death of hSOD1G93A animals (which are predisposed toward death) by 13 days and extended the average life span by 9 days (Fig. 6B). However, MSCs-Ngn1 did not alter the final endpoint, and all animals ultimately died with the average life span 19.3 ± 0.2 weeks. It is probably due to that we utilized the hSOD1G93A animals with the high copy numbers and maintained the animals without loss of copy numbers by breeding male transgenic animals to naive B6SJLF1/J dams. It is well known that the life span of SOD1 mice varies a lot in proportion to the copy numbers (1). The original hSOD1 mutant mice with high copy numbers ranging from 22 to 28 copies were known to survive for 129 days (22,23), which is quite similar to our animals. Interestingly, unprocessed MSCs induced a more prominent delay in disease onset when administered as two injections at 13 and 15 weeks than when delivered as a single injection, as evidenced by the significant enhancement in rotarod and PaGE test scores compared to the PBS-treated group at 15 and 16 weeks (compare Figs. 6 and 7). These results, which were similar to those produced by a single injection of MSCs-Ngn1, suggest that neural induction of MSCs may substitute for multiple transplantations of unprocessed MSCs in the treatment of ALS patients.
The most general method for CNS targeted stem cell delivery is direct injection into the brain parenchyma. Unfortunately, local transplantation may limit therapeutic effects in ALS, because degeneration of motor neurons occurs in a diffuse manner in the spinal cord, motor cortex, and brainstem nuclei of ALS patients; thus, direct application of therapeutic cells into the spinal cord may be ineffective in attenuating disease progression (40). By comparison, intravenous delivery of stem cells can effect on any part of widespread degenerative tissue. The ability of transplanted stem cells to migrate to the site of damaged tissue has been confirmed in various neurological diseases, including traumatic (34,36) or ischemic brain injuries (27,28,62), spinal cord injury (4,15,26,56), and brain tumor (48). Therefore, it was expected that the intravenous route of stem cell administration in the treatment of ALS would be more effective than the intraparenchymal route. In this study, we showed that injected MSCs-Ngn1 cells were detected within the CNS 2 and 8 weeks after intravenous transplantation (Figs. 2 and 4), suggesting an active migration of these cells from the periphery into brain and spinal cord.
Although details of the mechanism are still poorly understood, migration of MSCs to the injured sites involves various cytokines and chemokine receptors. In the brainstem and spinal cord of both ALS patients and animal models, activated microglia, astrocytes, and T lymphocytes, which are capable of secreting numerous cytokines and chemokines (10,16), are increased in numbers (2,25,41). We found that MCP-1 expression was markedly increased in hSOD1G93A mice whereas SDF-1 expression was constitutively high (Fig. 3A). The enhanced levels of MCP-1 in the spinal cord of these ALS mice could be due to breakdown of the blood–spinal cord barrier and microhemorrhages prior to motor neuron degeneration (74). We also found that both unprocessed MSCs and MSCs-Ngn1 express receptors for these cytokines. Collectively, these observations suggest that unprocessed MSCs and Ngn1-MSCs in the circulation are able to migrate to the CNS in response to a heightened immune response and/or as a consequence of the breakdown of blood vessels in the region where motor neurons degenerate. Indeed large numbers of injected cells were present 2 weeks after transplantation in animals treated with MSCs-Ngn1, whereas the counts were lower in animals treated with unprocessed MSCs (Fig. 2). More MSCs-Ngn1 cells populated the spinal cord parenchyma 8 weeks after the transplantation (Fig. 4C, D). In those periods, the animals with MSC-Ngn1 showed higher performance in motor functions, although the overall survival was not enhanced. Under the culture conditions employed, we observed no significant differences in the expression levels of neurotrophic factors such as BDNF, GDNF, VEGF, and HGF between MSCs and MSCs-Ngn1 (data not shown). Taken together, these results indicate that the increase in the number of MSCs-Ngn1 cells is primarily due to greater tropism to the CNS. However, the mechanism underlying the enhanced tropism will require further study.
ALS is a progressive disease; thus, establishing the appropriate timing of transplantation and conferring a long-lasting benefit will be crucial for clinical efficacy. In this study, transplantation of Ngn1-MSCs at preonset ages (8 weeks) delayed disease onset and moderately improved motor functions but did not significantly delay disease progression or extend life span. We demonstrated that transplantation of Ngn1-MSCs near the disease onset was more effective than transplantation at preonset ages (compare Figs. 5–7). One possible explanation for this observation is that the tropism of systemically transplanted cells toward the CNS requires inflammatory cues, as discussed above. This may be the reason that transplantation of Ngn1-MSCs at disease-onset is more effective than pre-onset transplantation. By comparison, earlier studies with hNT neuronal cells showed that intraspinal transplantation at a pre-onset stage (7–8 weeks) delayed the onset of motor deficits and increased the survival of recipient mice (20,66), whereas transplantation during the disease-progression stage (16 weeks) failed to extend animal survival (19). Taken together, these observations indicate that therapeutic efficacy may not only depend on transplantation timing, but also on cell type (e.g., stably transformed neuronal hNT cells vs. neurally induced Ngn1-MSCs) and delivery route (intraspinal vs. intravenous).
In many studies, different doses of stem cells were transplanted. The first report by Garbuzova-Davis et al. on cell dose for ALS with hNT neurons showed that high-dose transplants (390,000 cells) did not provide increased benefits compared to low-dose transplants (130,000 cells) (21). In another report, this same group administered three different doses (10 × 1 0 6, 25 × 1 0 6, and 50 × 1 0 6) of mononuclear human umbilical cord blood cells into pre symptomatic hSOD1G93A mice. Their results showed that a cell dose of 25 × 1 0 6 cells had the most beneficial effect on many therapeutic parameters (18). Until now, there has been no consensus on whether there is a correlation between transplanted cell number and therapeutic effects. In our study, the transplanted cell dose was increased by repeated transplantation, and this strategy showed remarkable efficacy compared to a single dose (compare Figs. 6 and 7). Our results have much in common with a previous study by Zhang et al (71). In that study, triple injection of MSCs into the cerebrospinal fluid yielded significant therapeutic effects, including attenuated weight loss, enhanced motor performance, decreased motor neuron loss, and increased survival in hSOD1G93A mice, whereas a single transplantation did not affect disease progression. Based on these combined results, we conclude that the multiple-injection strategy provide a more sustained therapeutic effects compared to a single dose, also MSCs are a more suitable cell source for repeated transplantation than are other allogeneic cell sources.
In summary, we have shown that neural induction with Ngn1 enhances the therapeutic functions of MSCs with respect to delaying disease progression and prolonging life span in hSOD1G93A mice. Our study identified several critical parameters that might affect the outcome of future clinical studies, including the role of neural induction, the number of treatments, and transplantation timing. Finally, our results suggest that MSCs with neural properties are more suitable for the long-term treatment of ALS than are unprocessed MSCs or other allogeneic cells of neural origin.
Footnotes
Acknowledgments
This work was partly supported by Brain Research Center of the 21st Century Frontier Research Program (2011k000262) and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Education, Science and Technology (2010-0020406) to HSK and by Basic Science Research Program through NRF funded by the Ministry of Education, Science and Technology (2010-0023676) to SSK. Conflicts of Interest: The authors declare no financial or other competing interests.