
Abstract
Transplanted embryonic stem (ES) cells, following myocardial infarction (MI), contribute to limited cardiac repair and regeneration with improved function. Therefore, novel strategies are still needed to understand the effects of genetically modified transplanted stem cells on cardiac remodeling. The present study evaluates whether transplanted mouse ES cells overexpressing TIMP-1, an antiapoptotic and antifibrotic protein, can enhance cardiac myocyte differentiation, inhibit native cardiac myocyte apoptosis, reduce fibrosis, and improve cardiac function in the infarcted myocardium. MI was produced in C57BL/6 mice by coronary artery ligation. TIMP-1-ES cells, ES cells, or culture medium (control) were transplanted into the peri-infarct region of the heart. Immunofluorescence, TUNEL staining, caspase-3 activity, ELISAs, histology, and echocardiography were used to identify newly differentiated cardiac myocytes and assess apoptosis, fibrosis, and heart function. Two weeks post-MI, significantly (p < 0.05) enhanced engraftment and cardiac myocyte differentiation was observed in TIMP-1-ES cell-transplanted hearts compared with hearts transplanted with ES cells and control. Hearts transplanted with TIMP-1-ES cells demonstrated a reduction in apoptosis as well as an increase (p < 0.05) in p-Akt activity compared with ES cells or culture media controls. Infarct size and interstitial and vascular fibrosis were significantly (p < 0.05) decreased in the TIMP-1-ES cell group compared to controls. Furthermore, MMP-9, a key profibrotic protein, was significantly (p < 0.01) reduced following TIMP-1-ES cell transplantation. Echocardiography data showed fractional shortening and ejection fraction were significantly (p < 0.05) improved in the TIMP-1-ES cell group compared with respective controls. Our data suggest that transplanted ES cells overexpressing TIMP-1 attenuate adverse myocardial remodeling and improve cardiac function compared with ES cells that may have therapeutic potential in regenerative medicine.
Introduction
Cell transplantation strategies using renewable sources of cardiac myocytes to repair damaged myocardium, replace lost cardiac cells, and prevent remodeling following myocardial infarction (MI) has gained significant attention. We, as well as others, have shown that transplanted embryonic stem (ES) cells post-MI have the potential to engraft, differentiate into cardiac-specific cell types, limit adverse myocardial remodeling, and improve heart function (3,14,33,34). However, inhibition of adverse cardiac remodeling following cell transplantation is minimal relative to normal heart architecture and overall cardiac function. Therefore, novel approaches are needed to augment the efficacy by which transplanted ES cells enhance cardiac repair in order to optimize cardiac regain of function following cell transplantation therapy.
Following myocardial infarction, ventricular architectural remodeling consequent to MI is characterized by various physiological and cellular changes including cardiac cell death via apoptosis and necrosis, fibrosis, and hypertrophy leading to end stage heart failure (2,15,19,25,34). In response to myocardial cell death post-MI, increased expression of extracellular matrix (ECM) proteins, specifically collagen types I and III, occurs evoking fibrosis formation (23,40). Fibrosis within the injured myocardium causes stiffening of the heart and overall systolic and diastolic dysfunction. Matrix metalloproteinases (MMPs) are a family of endopeptidases, which play a fundamental role in the degradation and aberrant production of ECM proteins leading to alterations in ventricular architect post-MI. Tissue inhibitors of metalloproteinases (TIMPs) are innate protease inhibitors of MMPs. TIMP-1, the most extensively characterized TIMP, is not only a well-documented antifibrotic protein (6,7,12) but also has recently been shown to exhibit antiapoptotic characteristics in noncardiac cell types such as breast epithelial cells (22,35,44). Moreover, we were the first to report that TIMP-1 is a cytoprotective released factor from ES cells, which prevents H2O2-induced apoptosis in vitro in cardiomyoblasts (H9c2 cells) (35). However, whether TIMP-1, when overexpressed in transplanted ES cells, can impact adverse cardiac remodeling post-MI by blunting host myocardium apoptosis and fibrosis has yet to be delineated.
In the present study, we hypothesize that ES cells overexpressing TIMP-1 (TIMP-1-ES cells), following transplantation into the infarcted myocardium, will attenuate apoptosis and fibrosis along with improving cardiac function. To accomplish this hypothesis, we generated ES cells overexpressing TIMP-1 (TIMP-1-ES cells), transplanted them into the infarcted myocardium, and evaluated their impact on engraftment, cardiac myocyte differentiation, apoptosis, fibrosis, and associated cardiac function. Our data show that TIMP-1-ES cells engraft and differentiate into cardiac mycoytes in the infarcted myocardium. We also suggest transplanted TIMP-1-ES cells post-MI significantly inhibit cardiac myocyte apoptosis compared with ES cells and the antiapoptotic effect of TIMP-1 is, in part, mediated through the Akt pathway. Furthermore, we demonstrate that transplanted TIMP-1-ES cells inhibit both interstitial and vascular fibrosis in the infarcted myocardium. Finally, we show that transplantation of TIMP-1-ES cells post-MI significantly enhances cardiac function.
Materials and Methods
ES Cell Culture
CGR8 ES cells with or without TIMP-1 were cultured and maintained on 0.1% gelatin-coated tissue culture plates in Dulbecco's minimum essential medium (DMEM) growth media supplemented with leukemia inhibitory factor (LIF), sodium pyruvate, glutamine, penicillin/streptomycin, nonessential amino acids, β- mercaptoethanol, and 15% ES qualified fetal bovine serum as we previously reported (38).
TIMP-1-ES Cell Generation
The mammalian expression vector (pTurboFP635-C) encoding red fluorescent protein (RFP) was purchased from Evrogen (Moscow, Russia). The cDNAs coding mouse TIMP-1 gene were generated by PCR cloning using high-fidelity DNA polymerase and PCR primers (forward, 5′-AGAGCAGATACCATGATG-3′ and reverse, 5′-GAAGGCTTCAGGTCATCG-3′). The PCR product was cloned to the Bgl II and BamH l site of the pTurboFP635 C-terminus as a single open reading frame to code for a TIMP-1/RFP fusion protein. The expression vector contained a neomycin resistance gene for selection of stable transfectants in CGR8 ES cell lines. The sequence of mouse TIMP-1 gene was confirmed by restriction enzyme digestion and DNA sequencing analysis.
CGR8 mouse ES cells were transfected with the TIMP-1 expression vector using Lipofectamine 2000 (Invitrogen). Selection media containing 100 mg/ml of the selective antibiotic G418 (Invitrogen) was added 48 h posttransfection. Selection medium was changed every 48 h, and cells were maintained for 2–3 weeks. Thereafter, G418-resistant ES cell colonies (labeled TIMP-1-ES cells) were selected, expanded, and maintained in ES cell culture medium. TIMP-1-ES cell clones were confirmed with elevated TIMP-1 protein expression as compared to parental CGR8 ES cells through Western blot analysis using a primary antibody against TIMP-1 (sc-5538, Santa Cruz) followed by incubation with goat anti-rabbit IgG secondary antibody (sc-2004, Santa Cruz).
TIMP-1 ELISA
Levels of secreted TIMP-1 were measured using a Ray Bio® Mouse TIMP-1 ELISA kit (ELM-TIMP1-001) as per the manufacturer's instructions. In brief, 1 week posttransfection and selection, medium was collected from tissue culture dishes containing growing ES and TIMP-1-ES cells. Samples were added for 2.5 h to the precoated wells, washed and incubated with biotinylated antibody for an additional hour. Following washings, horse radish peroxidase (HRP)-conjugated streptavidin solution was added for 45 min followed by incubation with the substrate solution for 30 min. Immediately thereafter, a stop solution was added and the color intensity was measured at 450 nm. The values obtained from the ELISA were corrected for the total protein concentration of each sample as estimated by the Bradford assay.
MI and Cell Transplantation
All mice were maintained and used as approved by the institutional animal review board. C57BL/6 mice (8–10 weeks old) were divided into four study groups (n = 8 per group): Sham, MI + cell culture media (MI + CC), MI + ES cells, and MI + TIMP-1-ES cells. In brief, mice were anesthetized, endotracheally intubated, and ventilated using a rodent MiniVent (Harvard Apparatus). Anesthesia was maintained using 2.5% isoflurane throughout the procedure. Left thoracotomy was performed, and the descending left coronary artery was permanently ligated as we reported previously (18). Following ligation, two intramyocardial injections of 10 ml of medium containing 2.5 × 104 TIMP-1-ES cells or ES cells were delivered into the peri-infarct region. The chest was then closed, lungs expanded, and mice were weaned from the ventilator and extubated. Sham group animals received all identical surgical procedures with the exception of no coronary artery ligation. Mice received postoperative care, including analgesia (buprenorphine, 0.5 mg/kg), for 2 days or until normal behavior resumed. Day 1 (D1) and D14 following surgery, mice were sacrificed by pentobarbital (80 mg/kg) followed by cervical dislocation. Hearts were removed, transversely cut, and fixed in 4% paraformaldehyde for further evaluation.
Histopathology
Heart tissue was embedded and sectioned as previously described (34). In brief, heart tissue was embedded in paraffin blocks, cut in serial sections (5 mm), and placed onto Colorfrost Plus microscope slides (Fisher Scientific). Heart sections were deparaffinized using room temperature xylene followed by sequential incubation in 100%, 95%, and 70% ethanol for 3 min and washed with distilled water. Heart sections were then stained with Mason's trichrome. Left ventricular (LV) infarct size was quantified by measuring the total LV infarct area/LV area × 100%. To quantify interstitial fibrosis, infarct, peri-infarct, and noninfarct regions of the heart were examined, and fibrosis was quantified by measuring the total blue area/total heart area × 100%. Vascular fibrosis was quantified by measuring vascular fibrosis/ vessel area × 100% in one to two sections from five to eight hearts per group. All fibrosis measurements were made using NIH ImageJ software.
Immunohistochemistry
To quantify engrafted transplanted cells at D1 and D14 following MI, heart sections were deparaffinized as aforementioned and immunohistochemically stained using primary antibodies against RFP (AB232, Evrogen) and octamer-binding transcription protein 3/4 (Oct 3/4; sc-5279, Santa Cruz). Heart sections were then incubated with Alexa 488- or Alexa 568-conjugated secondary antibodies (Invitrogen), respectively. Sections were washed, air dried, and mounted with Antifade Vectashield mounting medium. Colocalization of RFP and Oct 3/4-positive cells were counted as engrafted transplanted cells in one to two heart sections from n = 6–8 animals/group. To quantify the amount of differentiated cardiac myocytes from transplanted cells at D14, hearts were immunohistochemically stained with primary antibodies against sarcomeric α-actin (A2172, Sigma) and RFP (AB232, Evrogen) followed by incubation with appropriate secondary antibodies. Cells positive for both sarcomeric α-actin and RFP were counted as newly differentiated cardiac myocytes derived from transplanted stem cells. Visualization of engrafted and differentiated cells was performed using Olympus fluorescent and Leica TSC SP2 laser scanning confocal microscopes.
Tunel and Cardiac α-Actin Staining
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed as previously described (36). In brief, heart sections were deparaffinized and permeabilized with proteinase K (25 μg/ml in 100 mM Tris–HCl). The TUNEL assay kit (TMR red; Roche Applied Biosystems) was used as per the manufacturer's instructions to detect host myocardium apoptotic nuclei. Apoptotic cells were colabeled with cardiac α-actin (A2172, Sigma) to detect apoptosis within cardiac myocytes. Heart sections were then mounted with Antifade Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) for nuclear staining and observed under Olympus and confocal microscopes: percentage of total apoptotic nuclei = (total number of red stained apoptotic nuclei)/(total number of blue stained nuclei with DAPI) × 100. Additionally, the percentage of apoptotic nuclei in cardiac myocytes was also determined by (total number of red stained apoptotic nuclei colocalized with sarcomeric α-actin)/(total number of blue stained nuclei with DAPI) × 100. Total apoptotic nuclei and apoptotic cardiac myocyte nuclei were quantitated in the infarct and peri-infarct regions of the heart in one to two sections from five to eight hearts from each group. These cell counts were performed as previously reported by us and others (9,13,37).
Caspase-3 Activity Assay
Caspase-3 activity was quantitated using a caspase-3 colorimetric activity assay kit (K106-200, BioVision). Hearts were homogenized in RIPA buffer containing protease inhibitor cocktail (Sigma), phenylmethylsulfonyl fluoride (PMSF) (1 mM), sodium orthovandate (2 mM), and sodium fluoride (5 mM), collected, and centrifuged at 11,766 rpms for 5 min. The supernatant was removed, and protein concentration estimation was performed using a Bio Rad assay. Manufacturer's instructions were followed to complete the activity assay. Developed blue color was measured in a microtiter plate reader (Bio Rad) at 405 nm. Caspase-3 activity graph was plotted as arbitrary units (A.U.).
Phosphorylated Akt Activity Assay
Phosphorylated Akt (p-Akt) was quantitated using a phospho-Akt1 (PAN) ELISA kit (X1844k, Exalpha Biological) as previously described and detailed in the instruction manual provided with the kit (37). In brief, hearts were homogenized, and proteins were estimated by the Bradford method. Samples (25 ml) and sample diluents (75 ml) were added to the wells labeled with captured antibodies provided in the kit for 2 h. Following washing, samples were incubated with detector antibody for 2 h, HRP conjugate for 30 min, and substrate for 30 min with washings in between each step. A stop solution was added and the developed yellow color was measured in a microtiter plate reader (Bio Rad) at 450 nm. Phospho-Akt graph was plotted as detailed in the kit.
MMP-9 Immunoassay
MMP-9 concentration was determined by an enzyme-linked immunoassay (MMPT90, R&D Systems). In brief, hearts from n = 8 animals/group were homogenized, and protein concentrations were estimated as aforementioned. Samples were loaded into monoclonal MMP-9-immobilized antibody-coated wells provided in the kit for 2 h. Following washings, a conjugate (polyclonal antibody for mouse MMP-9) was added for an additional 2 h. Again the wells were washed, and a substrate solution was added to each well for 30 min followed by the addition of a stop solution. A color reaction developed proportional to the amount of bound MMP-9 and was measured in a microtiter plate reader (Bio Rad) at 450 nm. MMP-9 graph was plotted as arbitrary units (A.U.).
Echocardiography
Cardiac function for all mice groups was assessed noninvasively by transthoracic echocardiography at D14. Mice were anesthetized with 2% isoflurane, placed and maintained on a temperature-controlled heating pad, and the chest was shaved. Echocardiography was performed using a Phillips Sonos 5500 Ultrasound system with a 15-6L hockey stick transducer. Left ventricular internal dimension-diastole (LVIDd), left ventricular internal dimension-systole (LVIDs), fractional shortening (LVIDd-LVIDs/LVIDd × 100), and eject ion fraction [left ventricular volume at end diastole (EDV) – left ventricular volume at end systole (ESV)/EDV] were calculated using the Teichholz formula (41) at the midpapillary muscle level in short-axis view.
Statistical Analysis
All values presented as a mean ± SEM. Statistical analysis was performed using the students' t-test, one-way analysis of variance (ANOVA), and the Bonferroni test. Statistical significance was assigned when p < 0.05.
Results
To elucidate the impact of TIMP-1 in transplanted ES cells post-MI on cardiac remodeling and function, we overexpressed TIMP-1 (Fig. 1A–C) in CGR8 ES cells. Following selection with G418, TIMP-1-ES cells were maintained in cell culture for 10–12 weeks. To determine stable transfection, Western blot analysis was performed, and our data shows a ~49 kDa fusion protein band representative of TIMP-1/RFP expression in TIMP-1-ES cells compared to parental ES cells (p < 0.001) (Fig. 1D). Additionally, to understand whether TIMP-1 is secreted in the conditioned media (CM) obtained from ES and TIMP-1-ES cells, a TIMP-1 ELISA was performed on ES-CM and TIMP-1-ES-CM. Our quantitative data show a dramatic change in secreted TIMP-1 in TIMP-1-ES-CM relative to ES-CM (p < 0.05) (Fig. 1E).

TIMP-1 vector and the generation of TIMP-1-ES cells. (A) Tissue inhibitor of metalloproteinases-1 (TIMP-1) cDNA was cloned into the pTurboFP635-C expression vector encoding red fluorescent protein (RFP). TIMP-1-ES (TIMP-1-embryonic stem) cells were selected following incubation with the selective antibiotic G418. Representative photomicrographs of TIMP-1-ES cells in bright field microscopy (B) and under fluorescence microscopy (C) to demonstrate RFP expression. (D) Representative photomicrographs of Western blot. Histogram shows significant overexpression of TIMP-1 in TIMP-1-ES cells relative to parental ES cells. *p < 0.001 versus ES cells. (E) Histogram shows quantitative analysis of secreted TIMP-1 in ES and TIMP-1-ES cells. *p < 0.05 versus ES cells. MCS, multiple cloning site.
Enhanced Engraftment and Cardiac Myocyte Differentiation in TIMP-1-ES Cell Transplanted Hearts
To assess the impact of TIMP-1 overexpression in transplanted ES cells on engraftment into the host myocardium, heart sections were double immunolabeled with antibodies against RFP to detect donor transplanted cells and Oct 3/4, a marker indicative of pluripotent stem cells (Fig. 2). Figure 2A and E shows Oct 3/4-positive cells in red, Figure 2F shows RFP-positive cells in green whereas no such cells are present in the MI + CC group (Fig. 2B), DAPI is shown in blue in Figure 2C and G, and merged images are depicted in Figure 2D and H. The boxed area in Figure 2H is enlarged to show colocalization of Oct 3/4, RFP, and DAPI in the TIMP-1-ES cell group. At D1, no significant difference was observed in the amount of engrafted transplanted ES or TIMP-1-ES cells (Fig. 2I). However, at D14 a dramatic difference in the amount of engrafted TIMP-1-ES cells was quantified relative to the amount of engrafted ES cells (p < 0.05) (Fig. 2J).

Enhanced engraftment of TIMP-1-ES cells in the infarcted myocardium. Representative photomicrographs showing heart sections stained with anti-octamer binding transcription protein 3/4 (Oct 3/4) for pluripotent cells in red (A and E), donor transplanted cells in green (B and F), nuclei stained with DAPI in blue (C and G), and the merged image (D and H). Scale bar: 20 mm. (I) Quantitative analysis of engrafted cells at D1 following myocardial infarction (MI). (J) Significant increase in engrafted TIMP-1-ES cells at D14. *p < 0.05 versus MI + ES cells. CC, cell culture media.
To quantify cardiac myocyte differentiation from transplanted ES and TIMP-1-ES cells, heart sections were double immunolabeled with antibodies against RFP and the cardiac myocyte marker, sarcomeric α-actin. Colocalization of RFP-positive cells with α-actin positive cells were counted as newly differentiated cardiac myocytes from transplanted stem cells (Fig. 3). Figure 3A, E, I, and M shows α-actin-positive cardiac myocytes (red), Figure 3J and N shows transplanted donor cells (green) whereas no such cells were present in the sham or MI + CC group (Fig. 3B, F), Figure 3C, G, K, and O shows total nuclei stained with DAPI (blue), and Figure 3D, H, L, and P shows merged images of triple-labeled sections depicting differentiated cardiac myocytes from donor transplanted cells. Upon quantification, we reveal TIMP-1-ES cell-transplanted hearts contain significantly more donor-differentiated cardiac myocytes compared to ES cell-transplanted hearts as depicted in Figure 3Q (p < 0.05).
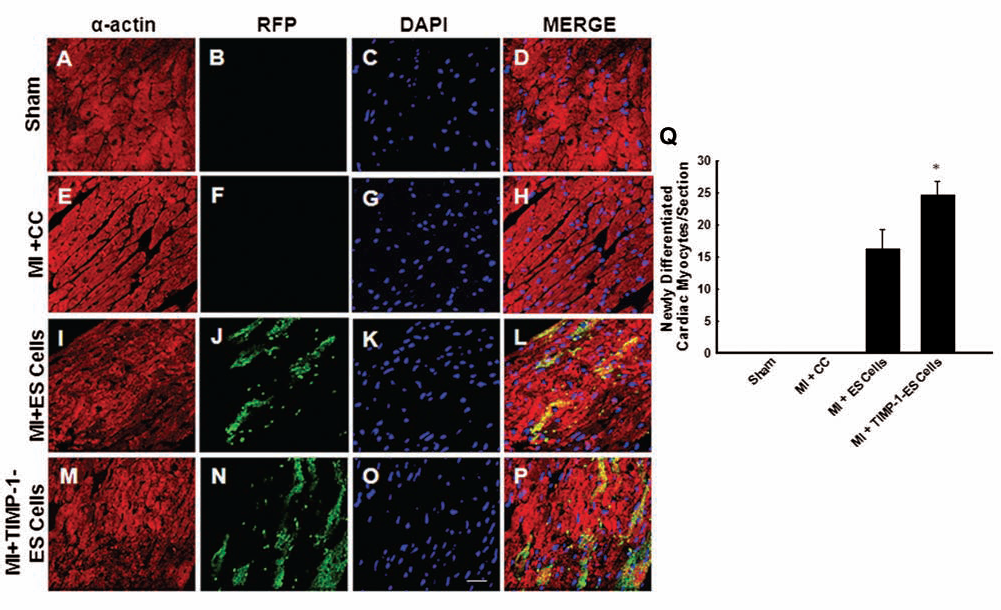
TIMP-1 enhances differentiation of ES cells into cardiac myocytes. Representative photomicrographs showing heart sections stained with anti-α-actin for cardiac myocytes in red (A, E, I, and M), anti-RFP for donor transplanted cells in green (B, F, J, and N), nuclei stained with DAPI in blue (C, G, K, and O), and the merged image (D, H, L, and P) showing coexpression of α-actin and RFP indicating differentiated donor cells. Scale bar: 20 mm. (Q) Quantitative analysis of differentiated donor ES cells into cardiac myocytes 2 weeks following MI. *p < 0.05 versus ES cells.
Reduced Infarct Region Following TIMP-1-ES Cell Transplantation
Representative whole-heart images depicting infarct size are represented in Figure 4A. Following quantification of infarct size, we reveal that the dramatic increase in infarct dimensions observed within the control MI + CC group was significantly blunted following transplantation of ES and TIMP-1-ES cells (p < 0.001) (Fig. 4B). Furthermore, our data demonstrate the extent of infarcted region within TIMP-1-ES cell transplanted hearts was significantly decreased compared to ES cell-transplanted hearts suggesting TIMP-1-ES cells are superior in reducing infarct size compared with ES cells (p < 0.01) (Fig. 4B).

TIMP-1 reduces infarct size following MI. (A) Representative photomicrographs of hearts demonstrating infarct size from C57BL/6 mice 2 weeks post-MI or sham surgery. Scale bar: 500 mm. (B) Histogram shows infarct size was significantly reducing following transplantation of TIMP-1-ES cells. *p < 0.001 versus MI + CC and #p < 0.001 versus MI + CC and MI + ES cells. LV = left ventricle.
Transplanted TIMP-1-ES Cells Prevent Apoptosis in the Infarcted Heart
To determine whether transplanted ES cells overexpressing TIMP-1 inhibit apoptosis compared to ES cells in the host myocardium, TUNEL staining was performed (Fig. 5). At D14, there was a significant decrease in TUNEL-positive nuclei post-MI in the infarct and peri-infarct regions of hearts transplanted with ES cells compared to controls (p < 0.001) (Fig. 5Q). Moreover, TIMP-1-ES cell-transplanted hearts contained significantly fewer total apoptotic nuclei compared to ES cell and cell culture medium groups (mean ± SEM; MI + TIMP-1-ES cells: 0.63 ± 0.06% vs. MI + ES cells: 0.94 ± 0.07% and MI + CC: 1.55 ± 0.12%, TUNEL positive/total nuclei, p < 0.05) (Fig. 5Q). Additionally, heart sections were colabeled with sarcomeric α-actin to demonstrate and quantitate apoptosis within cardiac myocyte nuclei (Fig. 5M–P). Our data show apoptotic cardiac myocyte nuclei were significantly reduced in hearts transplanted with TIMP-1-ES cells compared with ES cell and media-transplanted hearts (p < 0.05) (Fig. 5R). Next, to strengthen our TUNEL data, a caspase-3 colorimetric assay was performed on heart homogenates from each group. There was a significant decrease in caspase-3 activity in hearts treated with TIMP-1-ES cells post-MI compared to hearts transplanted with ES cells or medium alone (mean ± SE; MI + TIMP-1-ES cells: 171.75 ± 2.56 vs. MI + ES cells: 188.50 ± 1.80 and MI + CC: 203.00 ± 4.36, p < 0.05) (Fig. 6A).

Effects of transplanted ES cells overexpressing TIMP-1 on host cardiac myocyte apoptosis in C57BL/6 mice. ES and TIMP-1-ES cells were transplanted post-MI and hearts were examined for apoptosis 2 weeks following cell transplantation. Representative photomicrographs of total nuclei stained with DAPI in blue (A–D), apoptotic nuclei stained with Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) in red (E–H), and merged nuclei in pink (I–L). Scale bar: 100 mm. (M–P) Set of representative photomicrographs of section labeled with sarcomeric α-actin (M), apoptotic nuclei stained with TUNEL in red (N), total nuclei stained with DAPI in blue (O), and merged image (P) demonstrating apoptosis occurs within the cardiac myocyte. Scale bar: 50 mm. Boxed area in (O) enlarged to show colocalization of sacromeric α-actin, TUNEL-positive nucleus, and DAPI. (Q) Total apoptotic nuclei in the infarct and peri-infarct regions of the heart in one to two sections from five to eight hearts from each group. *p < 0.001 versus MI + CC, #p <0.05 versus MI + CC and MI + ES cells. (R) Quantitative cardiac myocyte apoptotic data. *p < 0.01 versus MI + CC, #p < 0.05 versus MI + CC and MI + ES cells.

Effects of TIMP-1-ES cell transplantation on caspase-3 and p-Akt expression. (A) Quantitative analysis of caspase-3 activity in hearts transplanted with and without stem cells. *p < 0.05 versus MI + CC, #p < 0.05 versus MI + CC and MI + ES cells. (B) Quantitative analysis of phospho-Akt activation. *p < 0.01 versus MI + CC, #p < 0.01 versus MI + CC and MI + ES cells.
TIMP-1 Enhances p-Akt Activation in Post-MI Hearts
Previous studies have shown that mechanisms of inhibited apoptosis involve the activation of cell survival pathways including p-Akt (38). Our data demonstrate that hearts transplanted with TIMP-1-ES cells did in fact have significant increase in the activation of p-Akt compared to hearts transplanted with ES cells and medium (mean ± SE; MI + TIMP-1-ES cells: 6.99 ± 0.22 vs. MI + ES cells: 5.75 ± 0.16 and MI + CC 4.61 ± 0.25, p < 0.05) (Fig. 6B), suggesting upregulation of cell survival pathways.
Reduced Fibrosis Following MI in TIMP-1-ES Cell Group (Fig. 7)

Effects of transplanted ES cells overexpressing TIMP-1 on interstitial and vascular fibrosis. Representative photomicrographs from sections stained with Masson's trichrome 2 weeks following coronary artery ligation demonstrating interstitial (A–D) and vascular (E–H) fibrosis. Scale bar: 100 mm. (I) Quantitative total interstitial fibrosis per section. *p < 0.01 versus MI + CC, #p < 0.05 versus MI + CC and MI + ES cells. (J) Quantitative analysis of vascular fibrosis in heart sections. *p < 0.001 versus MI + CC, #p < 0.01 versus MI + CC and MI + ES cells. VA, vessel area; VF, vessel fibrosis. (K) Average matrix metalloproteinase-9 (MMP-9) concentration within hearts from each group. *p < 0.01 versus MI + CC, #p < 0.05 versus MI + CC and MI + ES cells.
Interstitial fibrosis in the infarcted heart sections was measured at D14 and representative photomicrographs from each group are depicted in Figure 7A–D. Compared to medium alone, ES cell-transplanted hearts had a significant reduction in the amount of total interstitial fibrosis (p < 0.01) (Fig. 7I). Furthermore, the reduced interstitial fibrosis observed in TIMP-1-ES cell-transplanted hearts was significant when compared to ES cell and control hearts (mean ± SE; MI + TIMP-1-ES cell: 0.23 ± 0.03 mm2 vs. MI + ES cells: 0.47 ± 0.02 mm2 and MI + CC: 0.90 ± 0.00 mm2, p < 0.01) (Fig. 7I).
Next, quantitative assessment of vascular fibrosis (Fig. 7E–H) revealed ES cell-transplanted hearts had a significant decrease in the amount of vascular fibrosis compared to medium alone (p < 0.001) (Fig. 7J). Furthermore, the attenuation of vascular fibrosis in TIMP-1-ES cell-transplanted hearts reached statistical significance when compared to ES cells (p < 0.05) (Fig. 7J).
To determine whether inhibition of fibrosis was associated with changes in MMP-9, a well-documented MMP involved in the pathogenesis of MI, MMP-9 was quantified using an enzyme-linked immunoassay. Hearts transplanted with ES cells demonstrated significantly reduced MMP-9 concentration compared with control hearts (p < 0.01) (Fig. 7K). Moreover, MMP-9 concentration within TIMP-1-ES cell-transplanted hearts was significantly less compared to hearts transplanted with ES cells or media (mean ± SE; MI + TIMP-1-ES cells: 21.19 ± 1.27 vs. MI + ES cells: 41.64 ± 3.83 and MI + CC: 63.81 ± 8.18, p < 0.01) (Fig. 7K).
Overexpression of TIMP-1 Improves Cardiac Function Post-MI
To evaluate the cardiac functional consequences of transplanted TIMP-1-ES cells post-MI, two-dimensional echocardiography was performed. Obtained primary raw data are depicted in Figure 8A. Cumulative quantitative data revealed that, although a trend of decreased LVIDd was observed between TIMP-1-ES cell-transplanted mice and ES cell or CC-transplanted mice, statistical significance was not attained (Fig. 8B). Conversely, left ventricular dimensions during systole were significantly reduced following transplantation of TIMP-1-ES cells relative to mice transplanted with ES cells or CC (p < 0.05) (Fig. 8C). Cardiac function data at D14 further suggest mice receiving ES cell transplantation following MI had significantly improved fractional shortening compared to medium alone (p < 0.001) (Fig. 8D). Moreover, mice transplanted with TIMP-1-ES cells post-MI had significantly increased fractional shortening when compared to mice transplanted with ES cells and medium post-MI at D14 (mean ± SE; MI + TIMP-1-ES cells: 44.81 ± 0.70% versus MI + ES cells: 41.01 ± 0.61% and MI + CC: 31.30 ± 0.88%, p < 0.05) (Fig. 8D).
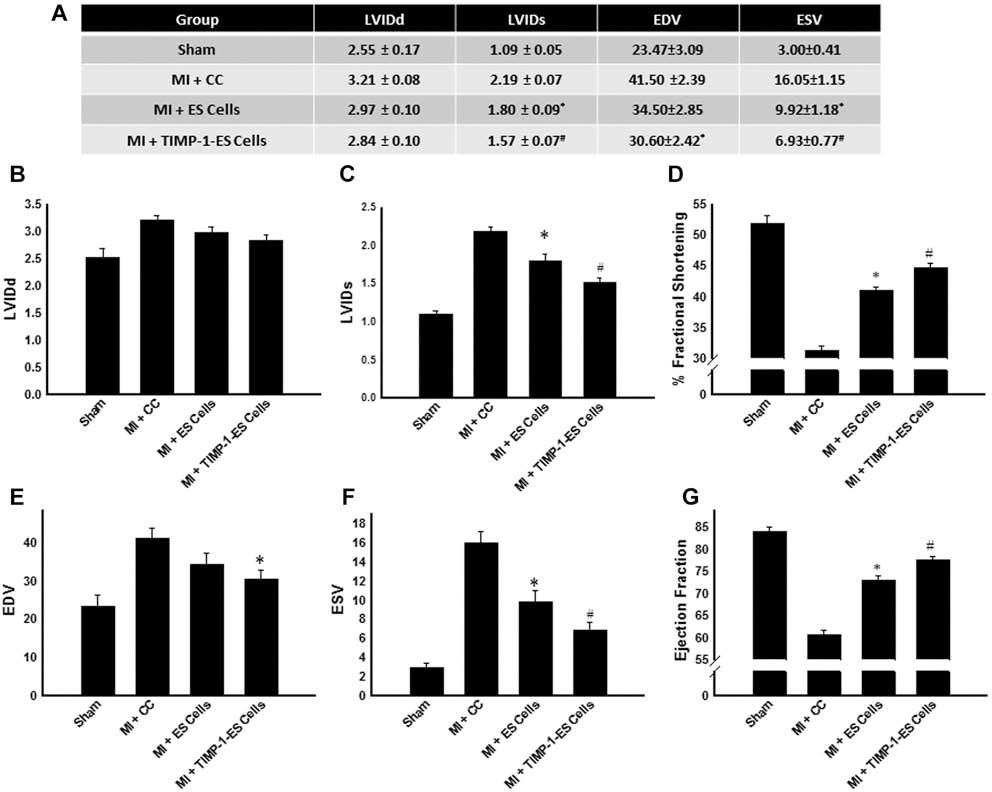
TIMP-1 overexpressed in ES cells improves cardiac function in C57BL/6 mice. Echocardiography was performed D14 following MI and fractional shortening and ejection fraction were quantified. (A) Primary raw data obtained from echocardiography analysis. (B) Average Left ventricular internal dimension-diastole (LVIDd). (C) Average LVIDs (systole). *p < 0.01 versus MI + CC and #p < 0.05 versus MI + CC and MI + ES cells. (D) Average fractional shortening at 2 weeks post-MI for all treatment groups. *p < 0.001 versus MI + CC, #p < 0.05 versus MI + CC and MI + ES cells. (E) Average EDV (left ventricular volume at end diastole). *p < 0.05 versus MI + CC. (F) Average ESV (left ventricular volume at end systole). *p < 0.001 versus MI + CC and #p < 0.05 versus MI + CC and MI + ES cells. (G) Quantified average ejection fraction. *p < 0.001 versus MI + CC, #p < 0.05 versus MI + CC and MI + ES cells. Data set is from n = 8 different animals/group.
End diastolic and systolic volumes are depicted in Figure 8E and F, respectively, for all study groups. As a dramatic reduction in ejection fraction was observed in MI + CC mice, this decrease was significantly blunted in mice transplanted with TIMP-1-ES and ES cells (p < 0.001) (Fig. 8G). Notably, mice transplanted with TIMP-1-ES cells additionally had significantly enhanced ejection fraction when compared with ES cell-transplanted mice (p < 0.05) (Fig. 8G).
Discussion
Many stem cell types examined thus far including skeletal myoblasts, bone marrow stem cells, bone marrow-derived hematopoietic stem cells, endogenous cardiac stem cells, mesenchymal stem cells, induced pluripotent stem (iPS) cells, and ES cells have shown improved cardiac function following transplantation post-MI (16,26,27,30,33). Improvement in cardiac function in past studies was demonstrated by newly differentiated cardiac myocytes and inhibition of apoptosis and fibrosis. However, observed effects of stem cell transplantation following MI was not enough to provide complete protection required to reestablish normal heart function. Therefore new strategies, such as modulation of ES cells using antiapoptotic and antifibrotic genes, are required. In the present study, we transfected ES cells with TIMP-1, an antiapoptotic and antifibrotic gene, transplanted them into the myocardium post-MI, and evaluated their effects on cardiac myocyte engraftment and differentiation, myocardial remodeling (apoptosis and fibrosis), and cardiac function.
Well-documented, endogenous TIMP-1 has been shown to be found within the extracellular matrix, membrane bound at the cell surface, and localized to the nucleus (22,31). The discriminating benefit propagated by constitutively active vector-driven TIMP-1 expression in ES cells is that TIMP-1 has the potential to exert its effects in an autocrine-mediated manner protecting transplanted ES cells from cell death or through paracrine mechanisms protecting the injured myocardium. We show that following transfection with the TIMP-1-RFP construct, the fusion proteins are abundantly expressed in and secreted by TIMP-1-ES cells. Previous studies have shown similar results indicating generated TIMP-1-GFP or TIMP-1-RFP fusion constructs are processed analogous to endogenous TIMP-1 (11,31).
The feasibility of exogenous transplanted cells to engraft into the infarcted myocardium is well documented (4,16,29,33). ES cells, derived from the inner cell mass of the blastocyst, are advantageous for nonautologous cell transplantation in that they confer minimal immunoreactivity consequent to the limited number to major histocompatibility complex surface proteins. Our study provides evidences indicating the enhanced survival of TIMP-1-ES cells in the injured myocardium following transplantation. Notably, these data for the first time suggest that a significant increase in TIMP-1-ES cells remain engrafted in the myocardium 2 weeks following transplantation contrary to the common observation that the majority of transplanted cells die within the first few days (5,32,48).
Regeneration of the infarcted heart is imperative to restoring the heart to pre-MI homeostasis. Intensive in vitro and in vivo research is underway using various factors to enhance and promote cardiac myocyte differentiation from stem cell populations (10,39,46,47). In this study, we have shown that our RFP-positive donor cells were elongated, nucleated, striated, and positive for sarcomeric α-actin. These characteristics of our RFP-positive cells indicate that not only do the transplanted cells survive in the injured myocardium but also differentiate into mature cardiac myocytes. Moreover, we have shown for the first time enhanced cardiac myocyte differentiation from transplanted ES cells post-MI by overexpression of TIMP-1. Our results are consistent with recent findings demonstrating the ability of TIMP-1 to promote oligodendrocyte, germinal center B cell, and UT-7 erythroid cell differentiation (8,17,28).
Next, we wanted to understand the effects of transplanted TIMP-1-ES cells on endogenous cardiac apoptosis. Well established, apoptosis is a major mechanism by which cardiac myocyte cell death occurs following MI leading to hypertrophy, fibrosis, and ultimately poor cardiac function. TIMP-1 has recently been identified as antiapoptotic in various cell lines including human osteosarcoma cells (MG-63), mouse bone marrow stromal cells (MBA-1), murine MC3T3-E1 osteoblasts, human breast epithelial cells, and rat cardiomyoblasts (H9c2) (24,35,43,44). The present study demonstrates a significant reduction in apoptosis in the host myocardium following transplantation with TIMP-1-ES cells compared to ES cells confirmed by TUNEL staining and caspase-3 activity assay. Our data are in accordance with a previously published study suggesting TIMP-1 conditioned media (CM) prevents H2O2-induced apoptosis in an in vitro model similar to ischemic myocardium (35).
Previous studies have reported a positive correlation between TIMP-1 and activation of the Akt signaling cascade-promoting breast epithelial and UT-7 erythroid cell survival (1,21). Our present study also provides evidence indicating hearts transplanted with TIMP-1-ES cells had significant activation of p-Akt compared to hearts transplanted with ES cells and medium controls. Although future detailed studies are necessitated to determine the exact mechanisms of the TIMP-1 antiapoptotic influence, our findings are in corroboration with previous studies suggesting mechanisms of inhibited apoptosis involve the activation of cell survival pathways including Akt (38).
Fibrosis, a major contributor to adverse cardiac remodeling, is consequent to increased MMP activation post-MI, which leads to ECM degradation, increased collagen deposition, stiffening of the heart, and ultimately poor cardiac function. In the present study, our cardiac fibrosis data (interstitial and vascular fibrosis) demonstrate significantly reduced fibrosis in hearts treated with TIMP-1-ES cells compared with hearts transplanted with ES cells. Additionally, hearts transplanted with TIMP-1-ES cells contained significantly less MMP-9 compared with ES cell and CC groups post-MI. These data are supported by previous studies indicating a reduction in MMP-9 expression in the presence of TIMP-1 overexpression (42). Following an MI, increased MMP activation is not paralleled by an increase in TIMP-1 expression (45). Therefore, we suggest significant reduction in the post-MI fibrosis observed in the TIMP-1-ES cell-transplanted hearts compared to ES cell-transplanted hearts may be attributable to inhibition of MMP-9 through overexpression of TIMP-1. However, further studies are required to define these mechanisms.
Finally, we needed to determine whether an increase in newly differentiated cardiac myocytes and inhibition of apoptosis and fibrosis in the host myocardium contribute to a decrease in infarct size and ultimately improve cardiac function following MI in TIMP-1-ES cell-transplanted animals. Infarcted myocardium contributes to aberrant left ventricular stiffness and diastolic dysfunction post-MI. Within the current study, we have shown a large infarct scar post-MI in the MI + CC control group. However, the infarct scar region was significantly diminished following TIMP-1-ES cell transplantation. Conceivably, we suggest that increased engraftment and differentiation following TIMP-1-ES cell transplantation repopulated the heart with additional functional cardiac myocytes, thereby reducing the infarct scar size. Moreover, in the present study, our data demonstrate that 2 weeks following MI, mice transplanted with TIMP-1-ES cells had significantly increased fractional shortening and ejection fraction compared to mice transplanted with ES cells or cell culture medium. We recognize that the mechanisms of improved cardiac function are complex and multifactorial. We do however suggest that the decrease in apoptosis and fibrosis, which is correlated with a decrease in infarct scar size, is directly related to the modulation of cardiac function observed within the current study. It is possible also to assert that improvement in ventricular function may be consequent to beneficial autocrine and paracrine mechanisms of TIMP-1 leading to continual differentiation of cardiac myocytes from transplanted TIMP-1-ES cells and protection of the host myocardium respectively leading to a decreased infarct size and ultimately cardiogenesis. These results are consistent with recent findings demonstrating regeneration of the infarcted myocardium following transplantation of ES cells (20,32,34). Findings from this study indicate that transplantation of TIMP-1-ES cells attenuate adverse cardiac remodeling and improve heart function following MI.
In conclusion, the major findings of the present study involving TIMP-1-overexpressing ES cells relative to ES cells include the following for the first time: (1) engraftment and differentiation into cardiac myocytes are significantly upregulated in transplanted TIMP-1-ES cells, (2) infarct size is significantly diminished in TIMP-1-ES cell-transplanted hearts, (3) TIMP-1-ES cells significantly inhibit cardiac apoptosis in the infarcted myocardium, (4) inhibition of apoptosis in hearts transplanted with TIMP-1-ES cells is mediated, in part, through the Akt pathway, (5) transplanted TIMP-1-ES cells significantly inhibit interstitial and vascular fibrosis as well as the profibrotic protein, MMP-9, following MI, and (6) transplanted TIMP-1-ES cells significantly improve overall cardiac function following MI through retention of functional cardiac myocytes and maintenance of the integrity of the host myocardium. However, future studies are required to identify further detailed mechanisms by which TIMP-1 prevents apoptosis and fibrosis in the infarcted myocardium. Additionally, as TIMP-1 is a secreted factor from ES cells, future studies are warranted to assess the role of TIMP-1-ES-CM in the infarcted myocardium, as previous studies have shown cytoprotective effects of CM in in vitro and in vivo models of ischemic myocardium (36,37).
Footnotes
Acknowledgments
The authors would like to thank Dr. Xilin Long for the generation of TIMP-1-ES cells, Reetu Singla for ES cell culture maintenance, Dr. Binbin Yan for assistance with confocal images, and Ajitha Dammalapati for histological assistance. This work was supported, in part, from grants from the National Institutes of Health (1R01HL090646-01 and 5R01HL094467-02 to D.K.S.). The authors declare no conflict of interest.