
Abstract
Human parthenogenetic stem cells (hpSCs) are pluripotent stem cells with enormous potential as cell sources for cell-based therapies: hpSCs may have histocompatibilty advantages over human embryonic stem cells (hESCs) and derivation of hpSCs does not require viable blastocyst destruction. For translation of all pluripotent stem cell-based therapies, derivation of differentiated cell products that are not contaminated with undifferentiated cells is a major technical roadblock. We report here a novel method to derive high-purity definitive endoderm (DE) from hpSCs, based on reproducing features of the normal human embryonic microenvironment. The method mimics the developmental process of transition through a primitive streak, using a differentiation device that incorporates a three-dimensional extracellular matrix (ECM) combined with a porous membrane. Treatment of undifferentiated hpSCs above the membrane results an epithelial-to-mesenchymal transition (EMT); thus, responsive cells acquire the ability to migrate through the membrane into the ECM, where they differentiate into DE. Importantly, the resultant DE is highly purified, and is not contaminated by undifferentiated cells, as assessed by OCT4 expression using immunocytochemistry and flow cytometry. The functional properties of the DE are also preserved by the process: DE differentiated in the device can generate a highly enriched population of hepatocyte-like cells (HLCs) characterized by expression of hepatic lineage markers, indocyanine green clearance, glycogen storage, cytochrome P450 activity, and engraftment in the liver after transplantation into immunodeficient mice. The method is broadly applicable and we obtained purified DE using hESCs, as well as several hpSC lines. The novel method described here represents a significant step toward the efficient generation of high-purity cells derived from DE, including hepatocytes and pancreatic endocrine cells, for use in regenerative medicine and drug discovery, as well as a platform for studying cell fate specification and behavior during development.
Keywords
Introduction
The first intentionally created human parthenogenetic stem cells (hpSCs) were derived from the inner cell mass of blastocysts of unfertilized oocytes activated by chemical stimuli (41). These hpSCs, like human embryonic stem cells (hESCs) (39,53), undergo extensive self-renewal and have pluripotential differentiation capacity in vitro and in vivo, giving rise to cells of all three germ layers. Using the spontaneous or chemical activation of an oocyte possessing the rare trait of human leukocyte antigen (HLA) homozygosity or alternate oocyte activation techniques yield hpSCs (heterozygous at most loci except HLA or HLA homozygous at all loci). hpSCs thus have clinically relevant histocompatibilty advantages in significant segments of the human population, due to HLA homozygosity (25,40,41,51). These hpSCs carrying common HLA haplotypes may reduce the risk of immune rejection (compared to hESC-derived cells) after transplantation of their differentiated derivatives into HLA-matched recipients. Moreover, creation of hpSCs is not associated with the ethical concerns associated with hESC derivation.
Two promising applications of pluripotent stem cells involve cell replacement therapy for diabetes (9,12) or chronic liver diseases (3,4,9,20,46). Production of high-purity definitive endoderm (DE) is a critical first step in the generation of therapeutically useful cells of the DE lineage, including hepatocytes and pancreatic endocrine cells.
DE is formed during gastrulation from epiblast cells that undergo an epithelial-to-mesenchymal transition (EMT) and ingress through the embryonic primitive streak (27,42). Upon differentiation signaling from the environment, epithelial-like cells of the epiblast undergo multiple morphologic and biochemical changes that enable them to assume a mesenchymal cell phenotype. That phenotype includes disruption of the intracellular adhesion complexes and loss of epithelial cell apical–basal polarity (2,31,37). These cytoskeletal changes are critical for cells to leave the epithelium and begin migration (29,32). The completion of the EMT is signaled by the migration of mesenchymal cells away from the epithelial layer of origin. Once formed, the primitive streak, acting via ingression, generates the mesendoderm, which subsequently separates to form the mesoderm and endoderm (19) (see Fig 1A). Thus, only cells that undergo the EMT participate in this migration and contribute to the final population of DE.

Cell migration during definitive endoderm (DE) differentiation, in vivo and in vitro. (A) In vivo: Schematic of cell migration through primitive streak during gastrulation. Epithelial-like cells of the epiblast (orange) undergo epithelial-to-mesenchymal transition (EMT) and acquire migration ability (green cells). These cells ingress through the primitive streak, replace hypoblast cells (yellow), then differentiate further to mesoderm and DE. (B) In vitro: Schematic of a 3D differentiation device that simulates migration through the primitive streak. Under differentiation signaling, pluripotent stem cells (orange) undergo EMT (green cells). These cells migrate through membrane pores into 3D extracellular matrix (ECM; yellow) and continue differentiation toward DE under high-level activin A signaling. Thus, differentiated cells are separated from undifferentiated cells by the membrane and a high-purity population of DE is differentiated and physically isolated. (C) Hematoxylin and eosin stain of a section of paraffin-embedded, 3D differentiation system demonstrates two compartments of cells in 3D differentiation system after of 3 days of differentiation, one population above and one below the membrane. (D) Immunofluorescent labeling of a section of paraffin-embedded, 3D differentiation system demonstrates identity of DE cells located below the membrane [(sex determining region Y) box 17 (SOX17)-positive nuclei, green] distinct from the mixture of differentiated and undifferentiated [octamer-binding transcription factor-4 (OCT4)-positive nuclei, red] cells located above the membrane. Sections were prepared after 3 days of DE differentiation.
In vitro, DE has been derived from hESCs (11,18,30), hpSCs (55), and human-induced pluripotent stem cells (hiPSCs) (36,47,49,59), using high-level activin A and Wnt3a signals to mimic signaling received by cells during ingress at the primitive streak (7,26,38,45). Knowledge about the major differentiation signals directing stem cells toward DE has not translated into methods to differentiate highly purified DE without undifferentiated cell contamination in the cultures (5,44,54,55). For clinical application, these residual undifferentiated cells are a major safety concern since they can generate teratomas. For example, 7 of 46 mice developed teratomas after injection of unpurified pancreatic cultures of DE derivatives generated from hESCs (23). Moreover, undifferentiated cells that remain from the first stages of differentiation may significantly reduce efficacy of whole differentiation procedure. One of the most advanced protocols to derive hepatocyte-like cells from hESCs resulted in an estimated efficiency of 18–26%, and enrichment of the differentiated hepatocytes required a flow cytometry step (yielding a population in which 55% of cells expressed albumin) (3).
The problem of cell purity of differentiated DE has been addressed by several groups, recognizing the importance of generating DE devoid of undifferentiated cells. The best result was achieved by defined medium containing high-dose activin A, bone morphogenetic protein-4 (BMP4), fibroblast growth factor-2 (FGF2), and a chemical inhibitor of phosphoinositide 3-kinase (PI3K); however, pluripotency markers such as octamer-binding transcription factor 4 (OCT4) and NANOG were detectable in the final differentiated cell product (54).
All previous studies used a two-dimensional (2D) culture system (monolayer cultures on a flat plastic dish) and did not provide a substrate to promote mesendoderm migration. 2D culture systems also cannot easily present a physiologically relevant 3D extracellular matrix (ECM) environment, which provides the crucial signals and substrate for migration during gastrulation. Some attempts to provide ECM signaling as part of protocols for derivation of DE point to the importance of ECM in these protocols (1), but results are far from optimized for efficiency and purity of derived DE.
We describe here a simple and novel 3D differentiation system that captures important features of the gastrulation stage embryo, utilizing soluble growth factors to induce differentiation, 3D ECM to promote cell–cell and cell–ECM interactions, and a physical path (pores) for promoting migration. We show that application of the system to various pluripotent cell lines produces high-purity DE without contamination of OCT4-positive cells, and the DE can be differentiated further into functional hepatocyte-like cells (HLCs). These studies are also the first demonstration of differentiation of highly enriched HLCs from hpSCs.
Materials and Methods
All hpSC lines used in this study were previously derived by our research group (40,41) and are owned by International Stem Cell Corporation. The hESC line WA09 was provided by Dr. Mike West. Informed consent was obtained for generation of the cell lines from the donors and was approved by independent ESCRO Committee of the University of California Irvine.
Cell Culture and Differentiation
Undifferentiated hpSCs and hESCs were grown on mouse embryo fibroblast feeder layers in KnockOut-DMEM/F12 supplemented with 15% KnockOut serum replacement, 0.05 mM nonessential amino acids, 2 mM Glutamax-I, penicillin/streptomycin, 55 μM 2-mercapthoethanol (all from Invitrogen), supplemented with 5 ng/ml recombinant human FGF-basic (PeproTech) and 20 ng/ml recombinant human activin A (rh-activin A; R&D Systems). Cultures were manually passaged and split at ratios of 1:4–1:6 every 5–7 days.
HepG2 cells (ATCC) were cultured in 3D ECM prepared with PureCol™ (Advanced BioMatrix) as described below, in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (HyClone).
For differentiation procedures, hpSCs or hESCs were plated at high density on top of the membrane of the differentiation device (see Fig. 1B). Control cultures were plated on flat plastic dishes (cell culture treated; Corning) pretreated with DMEM (Invitrogen) with 10% fetal bovine serum (FBS) (HyClone), and were cultivated for a further 2–5 days until the start of the differentiation procedure, in the hpSC growth medium described above.
The differentiation device was based on a 25-mm tissue culture insert (Nunc) with a synthetic membrane containing 8-μm pores (Whatman). For most experiments, a layer of 3D ECM was applied to the underside of the porous membrane.
The ECM was prepared on ice from a mixture of PureCol™ with 10× cell culture medium according to the manufacturer's instructions, with or without addition of human fibronectin (Sigma) to a final concentration of 100 μg fibronectin/ml ECM. (ECM with fibronectin was only used for cell migration assays.) To create a thin layer of 3D ECM, 200 μl of the iced ECM mixture was spread evenly on the underside of membrane of tissue culture inserts and incubated at 37°C for 60 min to induce gelation. The cell culture medium was added (overnight) to each insert containing 3D ECM before cell seeding.
Following published protocols (11,12) differentiation into DE was carried out in RPMI-1640 (Invitrogen) supplemented with Glutamax-I, penicillin/streptomycin, and 0.5 mg/ml human serum albumin (Sigma). For the first 24 h, this medium was supplemented with 100 ng/ml rh-activin A and 75 ng/ml recombinant mouse Wnt3a (R&D Systems). For the next 48 h, the medium was supplemented with 0.2% human AB serum (Fisher BioReagents) and 100 ng/ml rh-activin A. Wnt3a in combination with activin A increases the efficiency of mesendoderm specification, a bipotential precursor of DE and mesoderm, and improves the synchrony with which hESCs (12) and hpSCs (55) undergo DE formation.
To derive HLCs, DE cultures located in the 3D ECM of the differentiation device were cultivated for 3 or 5 days in KnockOut-DMEM/F12 supplemented with 20% KnockOut serum replacement, 30 ng/ml FGF4 (Pepro Tech), and 20 ng/ml BMP2 (PeproTech). Then cells were cultivated for 3 or 5 days in KnockOut-DMEM/F12 supplemented with 20% KnockOut serum replacement and 20 ng/ml hepatocyte growth factor (HGF; PeproTech) (instead of FGF4 and BMP2). Finally, the cells were cultivated for 5 days in hepatocyte culture medium (HCM; Lonza) supplemented with SingleQuots (Lonza), 20 ng/ml oncostatin M (R&D Systems), and 0.1 μM dexamethasone (Sigma).
All differentiation experiments were performed at least in triplicate. Graphical data error bars all represent standard deviations.
Cell Migration Assay
hpSCs and hESCs were plated on top of the membrane of the differentiation device, and put through the differentiation protocol described above. Cells were harvested at days 0, 1, and 2 of the differentiation procedure. The insert was washed gently in PBS and cells were removed from within the insert (on top of the membrane) using a dry cotton bud followed by two washes in PBS. To isolate intact cells embedded in the 3D ECM (or on the underside of the membrane in cases where the insert was used without the 3D ECM) the device was washed twice with PBS and incubated in 1000 U/ml collagenese solution (Invitrogen) at 37°C for 30 min. After incubation in collagenase solution the suspension of cell clumps was carefully collected from the bottom of the membrane and centrifuged. To obtain a single cell suspension the pellet was further dissociated using 0.05% trypsin (Invitrogen) at 37°C for 1–2 min, then centrifuged, resuspended in PBS with 3% FBS, and counted with a hemacytometer.
Immunostaining and Morphologic Staining
Cultures were fixed for 25 min at room temperature in 4% paraformaldehyde in PBS and permeabilized for 40 min in 0.1% Triton X-100 in PBS. Before immunostaining, the membrane with attached 3D ECM and embedded target cells were manually detached from the tissue culture insert. Antibodies and dilutions used in these studies are summarized in Table 1. The slides were mounted in Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories).
Source of Antibodies Used in Immunocytochemistry
To determine the histological and phenotypic characteristics of the migrating cells, the ECM-coated filter membranes of tissue culture inserts were cut out intact, fixed in formalin, embedded in paraffin, and sectioned (5 μm). Following deparaffinization and rehydration, the sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry in situ on the membrane, antigen retrieval was performed with citrate buffer (pH 6.0). Then the sections were costained with anti-Oct4 and anti-Sox17 [(sex determining region Y)-box 17] to distinguish undifferentiated hpSC from DE (Table 1). After labeling with the appropriate secondary antibodies, and nuclear counterstain with DAPI, the sections were captured using a Zeiss fluorescence microscope.
Real-Time Quantitative PCR (RT-qPCR)
Total RNA was isolated using the QIAsymphony automatic purification system, according to the manufacturer's instructions (Qiagen). Total RNA (100–500 ng) was used for reverse transcription with the iScript cDNA synthesis kit (Bio-Rad). PCR reactions were run in duplicate using 1/40th of the cDNA per reaction and 400 nM forward and reverse primers or the QuantiTect Primer Assay, together with Quantitest SYBR Green master mix (Qiagen). Real-time PCR was performed using the Rotor-Gene Q (Qiagen). Relative quantification was performed against a standard curve and quantified values were normalized against the input determined by one of the following housekeeping genes: cyclin G (CYCG), β-glucuronidase (GUSB), or TATA box binding protein (TBP). After normalization, the samples were plotted relative to the first sample in the data set and the standard deviation of the expression measurements was calculated. Primer sequences are reported in Table 2. RNA isolated from cryopreserved primary human hepatocytes (Invitrogen) was used as a comparison for gene expression of HLCs.
Real-Time PCR Primers

Flow Cytometry
Cells were dissociated using trypsin-like enzyme (TrypLE; Invitrogen) for 5 min, then pelleted and resuspended in PBS with 3% FBS. Labeling was carried out with CXC chemokine receptor 4-phycoerthyrin (CXCR4-PE; BD Biosciences), 10 μl/1 times; 106 cells for 30 min at room temperature. Isotype control was IgG2a, clone G155-178 (BD Biosciences). Cells were washed in buffer and resuspended in 1% paraformaldehyde (PFA). Samples were acquired on a Becton-Dickinson fluorescence activated cell sorter (FACS) Calibur 4-color flow cytometer and data analyzed using Becton-Dickinson CellQuest software. Data were gated using forward versus side scatter to eliminate debris and the resulting histograms plotted to reflect the mean fluorescence intensity of CXCR-4 versus the IgG2a isotype control.
For OCT4, α-fetoprotein (AFP), and α-1 antitrypsin (AAT) staining cells were fixed in 1% PFA in PBS for 1 h at room temperature. Permeabilization was performed for 30 min at room temperature in the permeabilization/wash buffer (R&D Systems). Antibody incubation was for 30 min at room temperature. Labeling was carried out with anti-ATT (Invitrogen), anti-AFP (DakoCytomation), or anti-Oct-4 Alexa Fluor®488 conjugate (Millipore).
Cellular Uptake and Release of ICG
Indocyanine green (ICG) is eliminated exclusively by hepatocytes, so uptake and elimination studies serve as a marker of hepatocyte maturity. ICG (1 mg/ml, Sigma) in DMEM was added to cell cultures (at late stage differentiation) and incubated at 37°C for 30 min. After washing, cellular uptake of ICG was documented using light microscopy. Cells were then returned to the culture medium and incubated for 6 h. The ICG was not detectable inside the cells 6.5 h after its addition to the cultures (58).
PAS Stain for Glycogen
Cultures of differentiated cells located in the 3D ECM were fixed with 4% paraformaldehyde (or Carnoy's fluid) and stained using a commercial Periodic acid-Schiff (PAS) staining system (Sigma) according to the manufacturer's instructions. Cultures of the cells treated with 0.5% diastase (Sigma) before PAS staining were used as a control.
PROD Assay
The pentoxyresorufin o-dealkylase (PROD) assay is a measure of cytochrome P450 CYP2B activity. Cultures of differentiated cells located in the 3D ECM were treated with phenobarbital sodium (Sigma) at a final concentration 1 mM for 72 h. The phenobarbital was then washed away, and replaced with medium containing the CYP2B substrate pentoxyresorufin (Sigma) at a concentration of 10 μM. After 20 min, living cell cultures were analyzed using fluorescence microscopy (24).
HLC Implantation in Mice
Animal studies were performed in compliance with institutional and NIH guidelines by Explora Labs (San Diego, CA). HLCs derived from hpSC line phESC-3 (41) were isolated from 3D ECM as described above and labeled with carboxyfluorescein diacetate, succinimidyl ester (CFSE) using the Cell Trace CFSE Cell Proliferation Kit (Invitrogen) according to the manufacturer's instructions. About 2 million cells in 50 μl Matrigel diluted 1:1 with HCM (or diluted Matrigel without cells) were injected into the spleen of the 4–6-week-old severe combined immunodeficient (SCID)-beige (Bg) female mice (Charles River). Experimental mice (n = 5) were injected with labeled cells and three animals from a control group received injection of Matrigel only. Forty-two days later mice were euthanized and the livers were either harvested for tissue sections or perfused to isolate hepatocytes. Liver sections were embedded in optimal cutting temperature compound (OCT; Tissue-TEK) and snap frozen until cryosectioning. Unfixed tissue sections were further analyzed using fluorescence microscopy for the presence of CFSE-positive cells, or fixed in 4% par-aformaldyde and analyzed for human albumin expression using immunohistochemistry (Table 1).
To collect hpSC-derived HLCs from the grafted animals, animals were anesthetized with ketamine/xylazine and the portal vein cannulated with a 24-gauge catheter (B Braun, Germany). The liver was perfused with Hanks' balanced salt solution (Sigma) supplemented with ethylene glycol tetraacetic acid (EGTA) (Sigma) for 3–4 min followed by collagenase IV solution (Sigma) for 5–6 min. Perfused livers were further teased apart with needles, resuspended in Leibovitz (L-15) medium (Sigma) supplemented with 10% FBS (Hyclone), and filtered through 100-μm cell strainers (BD). Isolated hepatocytes were washed twice in ice-cold L-15 medium supplemented with 10% FBS and analyzed by flow cytometry.
Results
Design of 3D Differentiation System
During vertebrate gastrulation, epiblast cells that have acquired the mesenchymal phenotype migrate through the primitive streak to form DE and mesoderm (13,50,56) (Fig. 1A). We built a differentiation device that separated DE from undifferentiated pluripotent stem cells based on similar migration behavior in vitro. (Fig. 1B–D). Essential features of the device are a membrane on which hpSCs can be cultured and segregated from cells that migrate through 8-μm pores, and 3D ECM on the underside of the membrane, through which differentiating cells can migrate and embed. Details of use of the device are described in detail in Materials and Methods.
Upon Undergoing EMT, Cells Acquire the Ability to Migrate Into 3D ECM
Under the imposed growth factors, ECM, and surface cues included in the differentiation protocol, the cells exhibited several hallmarks of EMT. Downregulation of junctional proteins is an essential part of EMT, and E-cadherin gene expression (Fig. 2A) in the differentiated cells was accompanied by loss of cell surface immunolocalization of the E-cadherin protein (Fig. 2B). N-cadherin is required for efficient migration and we showed upregulation of N-cadherin at the message (Fig. 2A) and protein levels (Fig. 2C) in differentiated cells.

Under differentiation signaling, pluripotent stem cells undergo an EMT and acquire ability to migrate. (A) RT-qPCR shows downregulation of E-cadherin and upregulation of N-cadherin expression during differentiation of human parthgenetic stem cells (hpSCs). d0 indicates results obtained from cells collected from above the porous membrane before induction of differentiation. d1, d2, d3 indicate results obtained from cells collected from the 3D ECM below the membrane, 24, 48, and 72 h after the start of the differentiation protocol. The y-axis indicates relative gene expression normalized to the d3 time point. Data in graphs is presented using SD error bars. (B) Immunofluorescent labeling of undifferentiated and differentiated cultures demonstrates presence of E-cadherin expression in undifferentiated cells before the application of differentiation signaling (0 h) and the lack of E-cadherin expression in cells collected from the 3D-ECM, 72 h after the start of the differentiation protocol (72 h). Image is uncoupled into green plus blue channels (E-cadherin and DAPI). (C) Immunofluorescent labeling of differentiated cultures demonstrates expression of N-cadherin in cells collected from the 3D ECM, 24 h after the start of the differentiation protocol. Image is uncoupled into green (N-cadherin, left) and green plus blue channels (N-cadherin and DAPI, right). (D) Phase contrast and indirect immunofluorescence microscopy demonstrate cytoskeletal rearrangements characteristic of cells undergoing EMT. Each image is shown in four versions: phase contrast (gray scale, far left), actin (red, middle left), paxillin (green, middle right), and superposition of actin, paxillin and DAPI (far right, DAPI in blue). Thirty-six hours after starting the differentiation protocol (36 h), actin stress fibers have replaced the cortical actin network present before differentiation (0 h), and the focal contact protein paxillin has relocalized from the cytoplasm to focal adhesions at the ends of the actin stress fibers. Actin cytoskeleton is visualized using Alexa Fluor® 546 conjugated phalloidin. (E) Migration assay: Vertical bars indicate numbers of cells collected below the porous membrane before differentiation (d0), 24 h (d1), and 48 h (d2) after the start of differentiation. Three different migration conditions are shown: membrane alone (“without 3D-extracellular matrix”), membrane with 3D ECM (“3D-extracellular matrix”), and membrane with 3D ECM supplemented by fibronectin (“3D-extracellular matrix with FN”). Data in graphs are presented using SD error bars. (F) Temporal dynamics of integrin expression during differentiation of stem cells into DE determined by RT-qPCR. The y-axis shows levels of relative gene expression. d0 through d3 indicate days from the start of differentiation. Data in graphs are presented using SD error bars.
Induction of EMT was also confirmed by characteristic structural rearrangement of the actin cytoskeleton. Undifferentiated stem cells had relatively few focal adhesions, a cortical arrangement of actin filaments, and a substantial cytoplasmic pool of paxillin (Fig. 2D). In cells that responded to the differentiation protocol, actin stress fibers replaced the cortical actin network and the focal contact protein paxillin relocalized from a mainly cytoplasmic distribution to a predominantly focal adhesion localization at the end of well-organized actin stress fibers (Fig. 2D). These structural rearrangements were accompanied by acquisition of another crucial behavior needed for EMT—the ability to migrate. We assessed the migration ability of undifferentiated and differentiated cells by following the migration of cells through 8-μm pores in the differentiation device. Before differentiation (day 0), no detectable numbers of cells were observed under the membrane, from the 0.6 million cells plated on top of the membrane. Under differentiation conditions, the number of cells detected under the membrane increased daily. By day 2 of differentiation about 0.5 million cells reached the underside of the membrane if ECM was not applied to the system. Application of 3D ECM to the underside of the membrane resulted in over 0.8 million migrated cells by day 2 (Fig. 2E). The 3D ECM used in these studies was predominantly collagen I. Since basal lamina contains fibronectin, we also tested a 3D ECM supplemented with fibronectin. With fibronectin, the number of cells detected in 3D ECM by day 2 of differentiation was 1.5-fold higher than in the system with 3D ECM that did not contain fibronectin, and was 2.7-fold higher than the system with membrane alone (Fig. 2E). Finally, by the end of DE differentiation, the system containing the membrane together with 3D ECM supplemented with fibronectin promoted quite good differentiation and migration efficacy: from 0.6 million undifferentiated hpSCs plated, more than 1.6 million cells migrated through the membrane by day 3. In contrast, a negative control experiment indicated that continued cultivation of hpSCs or hESCs using normal growth medium did not produce detectable numbers of cells below the porous membrane.
Under differentiation conditions, we also observed decreased expression of integrins, the cell surface receptors that mediate attachment of cells to the basal lamina (Fig. 2F). This result is consistent with the observation that the cells acquired expression patterns that weakened adherent junctions and facilitated active migration after undergoing EMT.
3D Differentiation System Allows Isolation of High-Purity DE
We characterized cells that migrated into the 3D ECM to determine their dynamic expression of DE-specific genes over the course of differentiation. Twenty-four hours after the start of differentiation, brachyury, a primitive streak marker, was expressed at high levels (Fig. 3A). Forkhead box A2 (FOXA2), cerberus 1, cysteine knot superfamily (CER1), and SOX17 transcripts, all associated with vertebrate DE, also exhibited a rapid increase in expression after the first 24 h. Expression of the chemokine receptor CXCR4 was delayed by 24 h relative to the other DE markers, but was detectable at 48 h. The expression of these four DE markers was maintained through day 3, but the high brachyury gene expression was transient, and suppressed by day 2. The pluripotency genes SOX2 and OCT4 were rapidly downregulated during the 3-day differentiation (Fig 3A). Thus, cells that migrated through the membrane into the 3D ECM demonstrated a temporal sequence of gene expression similar to that which occurs in the course of DE differentiation during vertebrate gastrulation.

3D differentiation system produces high-purity DE. (A) RT-qPCR shows temporal dynamics of marker gene expression during differentiation of stem cells into DE. The y-axis indicates relative gene expression in cells after migration and embedding in 3D ECM of the device (gray bars), or from a flat plastic dish (white bars). d0 indicates results obtained from cells collected from above the porous membrane or from flat plastic dish before the induction of differentiation. d1, d2, d3 data are from cells collected from 3D ECM below the membrane, or flat plastic dish, 24, 48, and 72 h after differentiation. Data in graphs are presented using SD error bars. (B) Immunofluorescence labeling demonstrates coexpression of SOX17 and brachyury (BRACH) during differentiation toward DE in the 3D differentiation system. After 24 h of differentiation (24 h), a majority of cells express brachyury (red). At 48 and 72 h, brachyury expression is undetectable and SOX17 expression (green) is increasing. At 36 h, the majority of cells express both proteins (orange and yellow shades), reflecting the transition of brachyury-positive precursors into SOX17-positive DE. (C) Flow cytometry analysis of DE derived in 2D (“flat plastic dish”) and 3D (“3D-extracellular matrix”) systems. Plots show numbers of cells versus fluorescence intensity, at day 3 of differentiation, for cells collected from the 3D ECM of the differentiation device or from a flat plastic dish. Cells were dissociated and stained with anti-CXC chemokine receptor 4 (CXCR4) antibody. Isotype-matched control antibody staining was performed using the same cells to determine background fluorescence. (D) Flow cytometric analysis demonstrates absence of OCT4-positive cells in the DE cultures collected from the 3D ECM of the differentiation device at day 3 of differentiation. Undifferentiated cells cultivated under conditions that support pluripotency are presented as positive control. Isotype-matched control antibody staining was performed using the same cells to determine background fluorescence.
Compared to cells differentiated in the same media in the 2D environment, the cells that migrated into 3D ECM showed more rapid kinetics of downregulation of pluripotency genes, significantly higher levels of endoderm gene expression (SOX17, FOXA2, CER1, CXCR4), higher peak levels of brachyury message at 24 h, and more rapid reduction of brachyury expression by 48 h (Fig. 3A).
No consistent increases in transcript levels associated with extraembryonic endoderm (SOX7, AFP), mesoderm [FOXF1, BMP4, mesenchyme homeobox 1 (MEOX1), fetal liver kinase-1 (FLK1)], ectoderm (SOX1, SOX2), or trophectoderm [human chorionic gonadotropin (HCG), caudal type homeobox 2 (CDX2)] was observed in cells embedded in 3D ECM by the end of activin A treatment.
In 2D differentiation DE paradigms, hpSCs as well as hESCs proceed through a gene expression sequence reminiscent of that occurring during gastrulation, as seen when pluripotent stem cells undergo an EMT coincident with initiation of brachyury expression, and SOX17-positive cells are derived from brachyury-positive precursors (11,55). To trace the origin of the SOX17-expressing cells in the population of cells that migrated into the 3D ECM, we characterized SOX17 and brachyury immunoreactivity over time. At 24 h there were a substantial number of brachyury-positive nuclei; by 36 h of differentiation more than half of the cells that expressed SOX17 were also brachyury immunoreactive, and at 48 and 72 h the majority of cells expressed SOX17 but brachyury protein was no longer detectable (Fig. 3B).
With the 3D differentiation system, we routinely observed that the overwhelming majority of cells in the 3D ECM were SOX17 positive by the end of activin A treatment, as determined by immunocytochemistry. To quantify the purity of the cell population, we performed flow cytometry analysis for the cell surface chemokine receptor CXCR4. By the end of activin A treatment, more than 90% of the cells in the 3D ECM were CXCR4 positive (Fig. 3C). In contrast, in a 2D system using the same differentiation protocol, about half the cells derived from hpSC were CXCR4 positive.
Recently published reports demonstrate that populations of DE that contain up to 80% CXCR4- or SOX17-positive cells can be derived from human pluripotent stem cells using conventional 2D culture system (5,44,54). These highly enriched DE cultures have significantly reduced OCT4 expression (four- to fivefold) compared to the original pluripotent cells, but undifferentiated OCT4-positive cells remain (5,44,54), a potential source of teratomas after transplantation (23). The problem of OCT4-positive cells that remain in the final differentiated cultures is even more significant for hpSCs using traditional 2D differentiation protocols: after a 3-day course of DE differentiation, 50% or more of the cells were OCT4 positive (55). Since undifferentiated cells (like epithelial cells) have limited ability to migrate, a major advantage of the membrane in our 3D differentiation system is that it serves to isolate undifferentiated OCT4-positive cells from the population of DE cells, confirmed by staining both cell populations on the differentiation device (Fig. 1C). Moreover, in the 3D differentiation system we observed more than 11-fold reduction in OCT4 gene expression in the differentiated cultures (Fig. 3A). These observations spurred us to determine the number of OCT4-positive cells in the population of DE generated using the 3D differentiation system. In three independent experiments we performed immunohistochemical staining of the differentiated cultures located on the underside of membrane using OCT4-specific antibodies; cultures of undifferentiated cells were used as a positive control. At least 3,000 nuclei were analyzed in each experiment. We did not observe a single OCT4-positive cell in the cultures isolated from the underside of the membrane in the 3D culture system. Absence of OCT4-positive cells in the final population of DE isolated from below the membrane by the end of day 3 of differentiation was confirmed by FACS analysis (Fig. 3D).
DE Derived in the 3D System Can Be Differentiated Into HLC
We tested the developmental competence of the derived DE cells by differentiating them further into HLCs. Following activin A treatment, differentiating cells were treated with FGF4 and BMP2, which support commitment of the ventral domain of the foregut to a liver cell fate (21,60). AFP and albumin gene expression became detectable on day 6 and increased continuously during the course of the differentiation procedure (Fig. 4A). AFP expression was not observed prior to day 5, as would be expected if substantial numbers of extraembryonic endoderm cells were present in the culture. By the end of FGF4 and BMP2 treatment, the morphology of the cells in the 3D ECM resembled the cuboidal shapes typical of hepatocytes (Fig. 4B). Moreover, the majority of the cells from this population expressed AFP, cytokeratin 18 (CK18), and hepatic nuclear factor 3β (HNF3β), detected by immunocytochemistry (Fig. 4C).
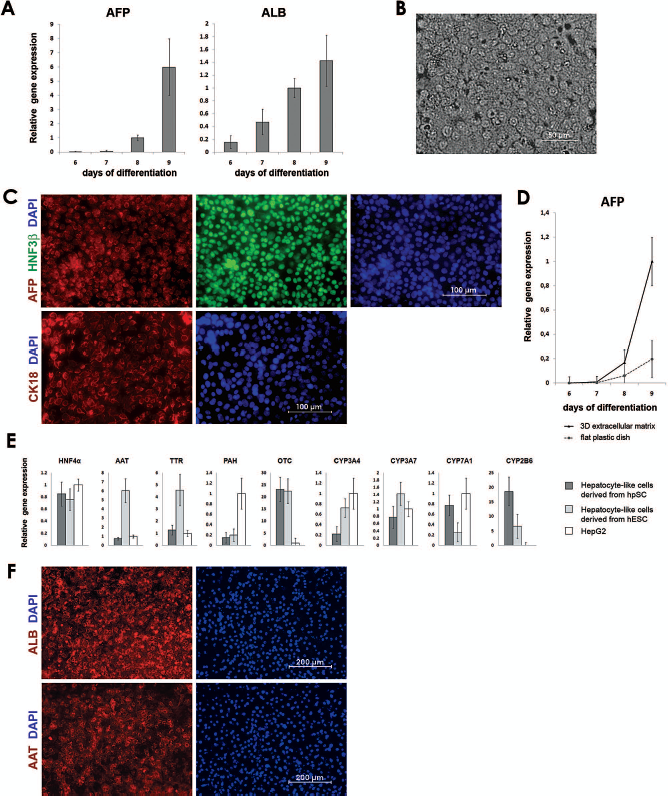
Characterization of hepatocyte-like cells (HLCs) derived from DE in the 3D differentiation system. (A) RT-qPCR demonstrates progressive upregulation of α-fetoprotein (AFP) and albumin (ALB) genes in cells collected from the 3D ECM during differentiation of DE toward HLCs. The y-axis indicates relative gene expression. Days of differentiation are counted from the start of the initial differentiation from pluripotent cells toward DE. Data in graphs are presented using SD error bars. (B) Phase contrast images show the cuboidal morphology of HLCs in the 3D ECM at day 8 of the differentiation protocol. (C) Immunofluorescent labeling of cells located in the 3D ECM demonstrates expression of early hepatocyte markers at day 8 of differentiation. (D) RT-qPCR shows increasing AFP gene expression during differentiation toward HLCs. AFP expression is greater in cells collected from the 3D ECM of the differentiation device (solid line) than from a flat plastic dish (dotted line). The y-axis indicates relative gene expression normalized to the d3 time point. Data in graphs are presented using SD error bars. (E) RT-qPCR demonstrates expression of hepatocyte markers at the end of differentiation toward HLCs. The y-axis indicates relative gene expression in cells collected from the 3D differentiation system (gray bars), normalized to that from the hepatic cell line HepG2 (white bars). Dark gray bars: HLCs derived from hpSC line phESC-3 (41); light gray bars: HLCs derived from human embryonic stem cell (hESC) line WA09. Data in graphs are presented using SD error bars. (F) Immunofluorescent labeling of cells located in the 3D ECM demonstrates expression of albumin (ALB) and α-1-antitrypsin (AAT) at the end of the differentiation protocol.
To promote the maturation of early hepatic cells derived in the 3D differentiation system, we used HGF treatment followed by oncostatin M and dexamethasone. Upon addition of HGF to the culture medium, differentiated cultures significantly increased AFP gene expression (Fig. 4D). This increase was more than five times higher in cells derived with the 3D system than in cells exposed to the same differentiation protocol in 2D. This observation may be a result of higher HLC purity of the 3D cultures and/or the possibility that cells cultivated in 3D ECM system express liver-specific proteins at higher levels than monolayer cells differentiated on a flat plastic dish.
The HLCs derived in the 3D differentiation system expressed a number of hepatic lineage genes including HNF4α, AAT, transthyretin (TTR), ornithine transacar-bamylase (OTC), and phenylalanine hydroxylase (PAH) (Fig. 4E, F). It is important to note that the levels of expression of these hepatocyte markers were similar in HLCs derived from hpSCs and HLCs derived from hESCs (Fig. 4E). These HLCs had functional characteristics of hepatocytes, including glycogen storage (shown by PAS staining in Fig. 5A) and uptake and elimination of IGC (Fig. 5B). A PROD assay demonstrated alkyloxyresorufin hydrolyzed to resorufin by hpSC-derived HLCs, confirming cytochrome CYP2B activity in the cells (Fig. 5C). Real-time quantitative PCR (RT-qPCR) also demonstrated CYP2B mRNA and three other P450 cytochromes, CYP3A7, CYP3A4, and CYP7A1 (Fig. 4E). To determine purity of the derived HLCs we performed flow cytometry of the cultures located in the 3D ECM and stained for specific hepatocyte markers. FACS analysis showed that the majority of cells express AFP and AAT; the channel increase over isotype control was 3.63-fold for AFP and 1.63-fold for AAT.

Characterization of HLCs derived from DE in the 3D differentiation system. (A) Periodic acid Schiff (PAS) staining (pink) indicates that the derived HLCs store glycogen. Nuclei were counterstained with hematoxylin (violet). (B) Green indicates indocyanine green (ICG) uptake by HLCs derived in the 3D differentiation system. (C) HLCs derived in the 3D differentiation system exhibit cytochrome P450 enzyme activity as evaluated by the pentoxyresorufin o-dealkylase (PROD) assay. Bright red in this merged fluorescence/phase contrast image indicates nonfluorescent alkoxyresorufin has been hydrolyzed to fluorescent resorufin by the P450 cytochrome CYP2B. (D) RT-qPCR demonstrates expression of hepatocyte markers at the end of differentiation toward HLCs. The y-axis indicates relative gene expression in cells collected from the 3D differentiation system (gray bars), normalized to those from human primary hepatocytes isolated from adult liver (white bars). Data in graphs are presented using SD error bars. (E) Flow cytometric analysis demonstrates the presence of carboxyfluorescin diacetate succinimidyl ester (CFSE)-positive cells in the population of cells isolated from mouse liver 42 days after transplantation of CFSE-labeled HLCs derived in 3D differentiation system (“HLC” plot). Population of cells isolated from the control liver (inoculated with a culture medium only) was analyzed to determine the background fluorescence. (F) Fluorescent microscopy analysis of frozen unfixed tissue sections demonstrates the presence of CFSE-positive viable cells in mouse liver 42 days after transplantation of CFSE-labeled HLCs derived in 3D differentiation system. (G) Immunofluorescent labeling of frozen tissue sections demonstrates the presence of cells expressing human albumin (ALB) in mouse liver 42 days after transplantation of HLCs derived in 3D differentiation system.
To estimate the maturation stage of the HLCs, we performed comparative analysis of the expression levels of genes associated with terminally differentiated primary adult human hepatocytes. As observed in Figure 5D, RT-qPCR analysis revealed expression of AFP (normally expressed in fetal, but not adult, hepatocytes), CYP1B1 and CYP1A1 (absent or present at very low levels in human adult liver), at significantly higher levels in comparison to human primary hepatocytes. HLCs expressed very little CYP2B6, CYP2D6, CYP3A4, and UDP glucuronosyltransferase 2 family, polypeptide B7 (UGT2B7), normally expressed in adult hepatocytes. Expression of TTR, another marker of hepatocytes, was maintained at the same levels in all tested cells (Fig. 5D). Overall, the results show that the cells are more fetal than adult in their expression profile. We observed that HLCs derived in our system continue to proliferate, with up to 14% of nuclei staining for Ki67 protein, a marker of the proliferating cells.
To assess the ability of derived HLCs to survive in vivo we transplanted CFSE-labeled cells into immunodeficient mice. Labeling with CFSE permits clear detection of transplanted cells, correlates with cell function, and permits the visualization of these cells by fluorescent microscopy (34,35). More than 40 days after transplantation, a significant population of CFSE cells was detected in mice liver by flow cytometry. Moreover, three solid peaks on FACS histograms demonstrate at least three successive generations of the inoculated HLCs (Fig. 5E), consistent with the proliferative phenotype. Clumps of viable CFSE-positive cells were also observed in sections of the host liver (Fig. 5F). Immunohistochemical analysis of these sections demonstrated presence of the cells expressing human albumin (Fig. 5G). These data indicate that HLCs derived from high-purity DE were able to migrate from the spleen, integrate into the liver, proliferate, and survive for at least 42 days.
3D Differentiation System Allows Derivation of High-Purity DE From Different Pluripotent Stem Cell Lines
We tested the 3D differentiation system on five different lines of human pluripotent stem cells, including one line of hESCs (WA09), and four lines of hpSCs [phESC-1, phESC-3, phESC-5 (41) and hpSC-Hhom-1 (40)]. The results reported here were obtained using phESC-3. However, all five stem cell lines gave similar results, including production of high-purity DE with up to 92% of cells positive for CXCR4, appropriate temporal dynamics of gene expression during differentiation to DE, expression of appropriate DE markers, and ability to differentiate further into HLCs that express hepatocyte markers and perform hepatocyte functions.
Discussion
These in vitro experiments were designed to reproduce conditions and microenvironment encountered by epiblast cells as they migrate through the primitive streak and differentiate into DE during embryonic development. We used the migration capacity of mesendoderm to isolate a high-purity population of DE differentiated from pluripotent hpSCs. The differentiation device was straightforward, with the critical arrangement of 3D ECM attached to the bottom of a porous membrane. Pluripotent stem cells (hpSCs or hESCs) were plated on top of the membrane, and exposed to soluble growth factors known to direct differentiation toward DE. The cells underwent EMT by gene expression, morphology, and behavioral criteria, and acquired migratory and invasive properties, as indicated by mass migration of differentiated cells through membrane pores into 3D ECM on the underside of membrane. The observed cell migration is very reminiscent of the physiological process that occurs during vertebrate gastrulation, when epiblast cells ingress through the primitive streak.
Both the porous membrane and the 3D ECM appear important to the improved performance of the differentiation device. The porous membrane was designed to exclude undifferentiated cells from the final cell population. We chose a pore size smaller to the diameter of an undifferentiated cell, so that the membrane would only be passable by cells that acquire the cytoskeletal changes necessary to migrate through the small opening, as part of EMT. The success of the design was supported by the nearly complete absence of cells below the membrane before cells were exposed to differentiation cues, even after extended periods of cultivation under pluripotency-maintaining conditions. Thus, the porous membrane contributed to the purity of the derived DE by excluding undifferentiated cells throughout the growth and differentiation paradigms.
Numerous reports suggest that the ECM plays a critical role in regulating stem cell differentiation into different lineages during embryonic development (6,8,14,16,22,33,48), including the differentiation associated with gastrulation. In our device, the 3D ECM may have enhanced the efficiency of cell differentiation in several ways. There may have been some direct (tropic) signaling from the ECM itself, promoting migration through the porous membrane, since the number of cells migrating increased when ECM was added to the system, and increased further still when fibronectin was added to the ECM. This finding is consistent with earlier reports that a collagen scaffold can be attractive for differentiating hepatic cells (1).
In addition, we speculate that the 3D cell distribution facilitated by the 3D ECM may promote cell–cell signaling that approximates the interactions among cells during gastrulation, a theoretical advantage of 3D over 2D systems. A 3D environment, in which each cell is surrounded by similar cells, may reinforce the chemical signals that each cell experiences from its neighbors, helping to synchronize and promote differentiation of the entire cell population. This supposition is consistent with our observation that, during differentiation to DE, characteristic changes in gene expression were greater in amplitude and narrower in time for the 3D system than for the 2D system. The supposition is also consistent with a growing literature showing that many cell types have different secretory profiles when cultured in 3D versus 2D (15).
Our results indicate that the cell type detected in the 3D ECM by the end of activin A treatment was authentic DE. Marker analysis at the protein and RNA levels was consistent with the formation of DE. Because brachyury expression has not been identified in the primitive endoderm lineage, the observation that SOX17 expression is initiated in brachyury-positive precursors, together with the absence of SOX7 and AFP expression, further strengthens the conclusion that the SOX17-positive cells were DE rather than primitive endoderm, which also can migrate (57). The purity of the derived DE is very high, with flow cytometry showing more than 90% of cells positive for CXCR4, for all stem cell lines investigated. In similar studies, using 2D systems the fraction of authentic DE cells is reportedly 50–80% for different hESC lines (11) and 50% or less for hpSCs (55).
Further directed differentiation of DE cells within the 3D ECM produced HLCs that stored glycogen, took up and eliminated IGC, and expressed active CYP2B enzyme. The cells also assumed the characteristic cuboidal shape of mature hepatocytes, and expressed a variety of hepatocyte genes and proteins, including four members of the P450 cytochrome family. These results indicate the differentiation competence of the DE cells, and the effectiveness of the 3D ECM as an environment for cell differentiation. The full repertoire of adult cytochromes would be necessary for use of these cells in toxicity studies, but the fetal hepatocyte phenotype may be useful for clinical transplantation in selected pediatric liver disease patients after further characterization. Human fetal hepatocyte transplantation is already practiced in selected pediatric populations and under clinical study for chronic liver diseases in adults (http://clinicaltrials.gov/ct2/show/NCT01013194).
A major conclusion from our study is that 3D differentiation conditions were superior to our own and others' 2D culture systems to generate pure populations of DE and for efficient HLC generation. During derivation of DE, the 3D system induced greater expression of characteristic endoderm genes, better defined temporal peaks in gene expression, and a much higher percentage of CXCR4-positive cells after activin A treatment. After further differentiation of DE to HLCs the vast majority of cells in the 3D system performed some hepatocyte functions, while the 2D system produced only isolated colonies of HLCs.
Our results may have several implications for future work in stem cell and developmental biology. First, with its ability to exclude some cell types that respond differentially to signaling, the 3D differentiation system may allow us to derive high-purity cell populations from a wide range of pluripotent stem cells. The consistent results across cell lines suggest that any pluripotent stem cell capable of responding to direct DE differentiation signaling will produce an isolated high-purity population of DE cells in the 3D differentiation device.
Second, the selectivity provided by migration through a porous membrane, along with the physiological conditions provided by the 3D ECM, may be useful in a wider range of applications, including isolation of various cell populations during differentiation of stem cells, isolation of primary cell cultures from different tissues, or research on cell migration and invasion, including cell ingress into the primitive streak. The membrane pore size and the composition of the 3D ECM can be varied to suit the application, but the basic technique should be applicable to any cell type that has migratory capacity, or to populations of cells with different migratory capacities.
Third, the composition of the ECM is an important variable in cell differentiation, and this component of the differentiation device deserves further optimization. Teratani et al. (52) found that hepatocytes derived from mouse embryonic stem cells are sensitive to ECM composition, and that type I collagen may be optimal for directing embryonic stem cells toward the hepatocyte lineage. In our differentiation device we used ECM containing type I collagen as the prevailing component. However, a different combination of ECM or other proteins in the device may be optimal for other cell types and differentiation processes.
Fourth, the high purity achieved with the 3D differentiation system may reduce the need for other isolation and purification methods such as FACS and magnetic cell sorting. The reduced stress on the cells may improve the yield and selectivity in any further differentiation toward a final cell lineage. The virtual absence of OCT4-positive cells in the DE is an important step in developing safe cell products from pluripotent stem cells.
Finally, our results may help to establish hpSCs as a useful source of starting materials for stem cell technologies. Parthenogenetic stem cells avoid some of the ethical questions associated with hESCs. They may also reduce immunosuppression requirements for cell-based therapies, since they can be produced with HLA homozygosity to be histocompatible with a large segment of the human population. Until recently, very little was known about the capacity of hpSCs for directed differentiation into desired cell lineages. Early studies of hpSCs only demonstrated their capacity for spontaneous differentiation in vitro and in vivo (25,28,40,41). Animal studies have shown that parthenogenetic pluripotent cells can differentiate into functional cells (10,43). Recently, we demonstrated the differentiation of hpSCs into high-purity retinal pigment epithelium (RPE) that expresses appropriate RPE markers and is phagocytic (17). The RPE differentiation combined with results in this report indicate that hpSCs can indeed be differentiated into high-purity, functional cell types.
Summary
We describe a new differentiation device that adapts basic tissue engineering concepts (scaffold with pores, signals from ECM, and culture in 3D) to improve the purity of differentiated cells from hpSC and other pluripotent cells. The combination of pores and 3D ECM, in the presence of appropriate soluble signals, induces migration and EMT, resulting in high-purity DE and HLCs from a range of human pluripotent stem cell lines. Furthermore, the device isolates nonmigratory undifferentiated pluripotent stem cells from the final cell product. These results also support the clinically relevant differentiation capacity of hpSCs. Finally, the protocols tested here may be useful in a wide range of applications focused on cell differentiation, and isolation of primary cells.
Footnotes
Acknowledgments
We would like to thank Dr. Mike West for providing hESC line WA09 for this research and Dr. William Avrin for help with writing. Dr. Jeffrey Fair in part of the work that is connected with hESC research was supported by NIH Award JHR R01HL082606.