
Abstract
Mesenchymal stem cells (MSCs) of mammals have been isolated from many tissues and are characterized by their aptitude to differentiate into bone, cartilage, and fat. Differentiation into cells of other lineages like skeletal muscle, tendon/ligament, nervous tissue, and epithelium has been attained with MSCs derived from some tissues. Whether such abilities are shared by MSCs of all tissues is unknown. We therefore compared for three human donors the myogenic properties of MSCs from adipose tissue (AT), bone marrow (BM), and synovial membrane (SM). Our data show that human MSCs derived from the three tissues differ in phenotype, proliferation capacity, and differentiation potential. The division rate of AT-derived MSCs (AT-MSCs) was distinctly higher than that of MSCs from the other two tissue sources. In addition, clear donor-specific differences in the long-term maintenance of MSC proliferation ability were observed. Although similar in their in vitro fusogenic capacity with murine myoblasts, MSCs of the three sources contributed to a different extent to skeletal muscle regeneration in vivo. Transplanting human AT-, BM-, or SM-MSCs previously transduced with a lentiviral vector encoding β-galactosidase into cardiotoxin-damaged tibialis anterior muscles (TAMs) of immunodeficient mice revealed that at 30 days after treatment the frequency of hybrid myofibers was highest in the TAMs treated with AT-MSCs. Our finding of human-specific β-spectrin and dystrophin in hybrid myofibers containing human nuclei argues for myogenic programming of MSCs in regenerating murine skeletal muscle. For the further development of MSC-based treatments of myopathies, AT-MSCs appear to be the best choice in view of their efficient contribution to myoregeneration, their high ex vivo expansion potential, and because their harvesting is less demanding than that of BM- or SM-MSCs.
Keywords
Introduction
Until recently, the widely held view was that human adult stem cells are committed and restricted to differentiate only into cell lineages of the tissue in which they reside. However, evidence for the presence of multipotent stem cells in the stroma of virtually all postnatal tissues is accumulating (17). The precise location and physiological role of these so-called mesenchymal stem cells (MSCs) are, so far, poorly defined. Recent findings suggest that MSCs represent activated progeny of pericytes that line the abluminal side of blood vessels throughout the body (4,12,16). The biological mechanisms directing the fate of MSCs are not known, but it is generally assumed that tissue-specific environmental factors are important.
MSCs, first isolated from bone marrow (BM) (29), are defined as plastic-adherent fibroblast-like cells with extensive proliferation capacity in culture and the ability to differentiate in vitro to adipocytes, chondrocytes, and osteoblasts (27,62). Cells meeting these characteristics have been isolated also from adipose tissue (AT) (88), dental pulp (35), lung (69), peripheral blood (51), skeletal muscle (84), skin (39), and synovial membrane (SM) (21) of human adults, from amniotic fluid (42), placenta (41), and umbilical cord (28), and from fetal BM, blood, liver, lung, and spleen (6,40).
MSCs derived from different tissues share a number of nonhematopoietic cell surface markers [i.e., CD29, CD44, CD73, CD90, CD105, and human lymphocyte antigen (HLA)-ABC] (16,18). They do differ, however, in their ex vivo expansion capacity, gene expression profile (1,26,53,56), and differentiation potential (49,59,60,85). The question whether these differences are intrinsic (i.e., are dictated by the tissue of origin) is addressed in the present study with particular emphasis on the myogenic properties of the MSCs.
The aptitude of MSCs to acquire skeletal muscle cell properties upon myogenic stimulation in culture and their contribution to myotube/fiber formation in vitro and in vivo have been extensively studied in the past decade. However, the findings are diverse. An example is the response of MSCs to the demethylating agent 5-azacytidine. Exposure of MSCs derived from rat BM (82), human menstrual blood (15), or human SM (21) to 5-azacytidine during culture stimulates myotube formation and the expression of skeletal muscle-specific markers. However, a similar response was not seen in MSCs isolated from fetal and adult human BM [(7) and our unpublished observations]. Likewise, supplementation of culture medium with proteins released by damaged muscle or cultured myoblasts induces myogenic differentiation of rat BM-MSCs and of fetal human MSCs obtained from blood or BM but not of adult human BM-or umbilical cord blood-derived MSCs (7,58,72).
The contribution of human MSCs to murine skeletal muscles during their regeneration in vivo has been reported. Rodriguez et al. (66) demonstrated extensive expression of human dystrophin (in up to 90% of myofibers) in tibialis anterior muscles (TAMs) of dystrophic (i.e., mdx) mice at 6 months after local injection of massively expanded (>200 population doublings) MSCs isolated from AT of young donors. Integration into regenerating myofibers, either directly or via satellite cell intermediates, also has been documented for MSCs isolated from AT (54), BM (23,75), and SM (22) of adult donors as well as from fetal BM (7). These studies were performed either in mdx mice or in artificially injured muscles of immunodeficient mice and generally yielded less hybrid myofibers than observed by Rodriguez et al. (66).
In the absence of a side-by-side comparison of the myogenic properties of MSCs from various tissues, it remains undecided whether the aforementioned differences are to be attributed to the tissue source of the MSCs, donor-specific parameters like age, gender, and health status, cell culture-specific variables like the extent of ex vivo cell proliferation and the cell culture conditions, and/or the read-out system(s) used (19,20,66).
To address this issue, we compared for three human adults properties of MSCs isolated from three different tissues using both in vitro and in vivo assay systems. The in vitro studies included a comparison of the long-term growth characteristics, telomerase activity, immunophenotype, skeletal muscle gene expression profile, multilineage differentiation capacity, and myogenic fusion properties of the cells. The participation of LacZ-tagged MSCs in myofiber regeneration was quantified in cardiotoxin (CTX)-injured TAMs of nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (23). Immunohistological detection in hybrid myofibers of human skeletal muscle proteins served to establish the in vivo myogenic differentiation potential of the MSCs.
Materials and Methods
Isolation and Culture of MSCs
MSCs were isolated from AT, BM, and SM of three patients (donor 1: male, 65 years old; donor 2: male, 69 years old; donor 3: female, 72 years old). Samples were surgical waste collected with informed consent during orthopedic surgery and according to the guidelines of the Leiden University Medical Center (LUMC; Leiden, the Netherlands).
The procedure for isolating MSCs from BM samples has been described previously (47). To isolate MSCs from AT and SM we slightly modified the protocols of Rodriguez et al. (66) and De Bari et al. (21), respectively. Briefly, small pieces of roughly 1.5 g of AT and SM were minced by scissors and digested in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Breda, the Netherlands) containing 100 U/ml penicillin (Invitrogen), 100 μg/ml streptomycin (Invitrogen), and 3 mg/ml collagenase type 1 (Worthington Biochemical Corporation, Lakewood, NJ) for 70 min at 37°C while shaking. Cells were then separated from the remaining tissue fragments by filtration through a 30-μm pore size nylon mesh filter (Miltenyi Biotec, Bergisch Gladbach, Germany), collected by centrifugation, and seeded in growth medium (GM) composed of DMEM, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS; Invitrogen) in plastic 25-cm2 cell culture flasks (CELLSTAR; Greiner Bio-One, Frickenhousen, Germany) and incubated at 37°C in humidified air with 10% CO2. Forty-eight hours after seeding, the culture supernatant was replaced by fresh medium to discard nonadherent cells and to allow expansion of the adherent cell fraction. When a confluency of 70% was reached, cells were detached using 0.05% trypsin (Invitrogen) and replated at a concentration of 3–5 times; 103 cells/cm2 (45). MSCs of each source at passage 2 were aliquoted (2–5 times; 105 cells per aliquot) and cryopreserved. For further expansion, MSCs were seeded at a density of 3–5 times; 103 cells/cm2 in GM. Basic fibroblast growth factor (FGF2; Sigma-Aldrich, Saint Louis, MO) was added to the culture medium only when indicated.
To compare the long-term growth characteristics of the MSCs from the different sources, cells of passage 2 were thawed and seeded in 25-cm2 cell culture flasks at a density of 2 times; 103 cells/cm2 in GM. Cells were replated when cultures reached 70–80% confluence at the same density as at culture initiation. After 50–100 days (4–6 passages), cultures were split and cells of each tissue source were grown in parallel in GM supplemented or not with FGF2 to a final concentration of 0.5 ng/ml.
Cell counts at every passage were used to calculate (i) number of population doublings (PDs) using the formula PDs = [ln(No/Ni)]/ln2, where No is the number of cells collected (output) and Ni is the number of cells seeded (input) at that passage, and (ii) population doubling time (PDT) by dividing the number of PDs during that passage by its duration in number of days. The average PDT of each cell population was calculated at the linear range of the growth curve. Cumulative population doublings (CPDs) were calculated by adding the number of PDs of each passage to the sum of PDs obtained in the previous passages. The CPDs were graphically depicted as a function of days in culture. The doubling time maintenance (DTM) is defined as the interval (days) between the moment of culture initiation (time 0) and the time point at which the PDT begins to increase.
Cell Lines
The cell lines HepG2 [human hepatocellular carcinoma cells; American Type Culture Collection (ATCC), Manassas, VA], VH10 (human foreskin fibroblasts) (46), HeLa (human cervical carcinoma cells; ATCC), and C2C12 (murine myoblasts; ATCC) were cultured in GM at 37°C in a humidified atmosphere with 10% CO2.
A line of immortalized human myoblasts originating from a healthy donor (LHCN-M2) (86) was cultured in 75-cm2 cell culture flasks (CELLSTAR; Greiner Bio-One) coated with 0.1 mg/ml bovine collagen type I (PureCol; Inamed, Santa Barbara, CA) in F-10 Nutrient Mixture (Ham), 1% GlutaMAX-I, 20% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen). Human immortalized myoblasts of a Duchene muscular dystrophy patient (iDMD myoblasts) were grown in culture medium as described previously (14). Both human myoblast lines were kept in a humidified air/5% CO2 atmosphere at 37°C.
Telomerase Activity Assay
Telomerase activity was determined in MSCs of early (3–5) and late (12–18) passages using the Telo TAGGG Telomerase polymerase chain reaction (PCR) enzyme-linked immunosorbent assay (ELISA) kit (Roche, Mannheim, Germany), following the manufacturer's instructions. Briefly, 2 times; 105 cells were lysed in 200 lysis buffer of which 2 μl was used for the telomeric repeat amplification protocol assay. The telomerase reaction products were then amplified in 30 PCR cycles using a TRIO-Thermoblock (Biometra, Goettingen, Germany). The biotinylated PCR products were semi-quantified by ELISA. Absorbance was measured at 450 nm. Human embryonic kidney 293 cell lysate (supplied by the manufacturer) served as positive control. DNase-free RNase-treated aliquots of each cell lysate provided internal negative controls. The net absorbance of each sample was calculated by subtracting the signal obtained in the presence of the DNase-free RNase from that acquired in its absence. Telomerase activity of individual cell samples was expressed as a percentage of the net absorbance of 293 cells. The average absorbance of the 293 cell samples and their controls (n = 4) was 2.282 and 0.083, respectively.
Immunophenotyping
MSCs of passage number 5–13 were immunophenotyped by flow cytometry as previously described (81). All data are from cells cultured in GM. Details about the antibodies that were used for these experiments are given in Table 1.
mMAbs Used for the Immunophenotypic Characterization of MSCs

BD, Becton Dickinson and Company; BL, BioLegend; CLB, Sanquin; D, Dako; MB, Miltenyi Biotec; RD, RD Systems; SC, Santa Cruz; PE, phycoerythrin; FITC, fluorescein isothiocyanate; HLA, human lymphocyte antigen; ALPL, alkaline phosphatase; SSEA, stage-specific embryonic antigen. All antibodies were used at the concentrations recommended by the suppliers.
Multilineage Differentiation in Culture
The MSCs used for the multilineage differentiation studies were of passages 3–6. Adipogenesis and osteogenesis were induced and evaluated as reported previously (45). Chondrogenic differentiation was performed essentially according to a previously described protocol (6). After fixation of the cell pellets in phosphate-buffered 4% formaldehyde (Mallinckrodt Baker, Phillipsburg, NJ) they were embedded in paraffin, cut in 8-μm-thick sections, and processed for immunohistochemistry as described (37) by using a mouse monoclonal antibody (mMAb) directed against human collagen type II (Millipore, Billerica, MA: clone COLL-II; IgG1; 1:50). Bound antibodies were visualized with 3,3′-diaminobenzidine tetrahydrochloride (Sigma-Aldrich). Sections were counterstained with hematoxylin and mounted with Pertex mounting medium (Histolab Products, Gothenburg, Sweden).
Myogenic differentiation was induced with medium composed of DMEM, 100 U/ml penicillin, 100 μg/ml streptomycin, and insulin-transferrin-selenium-G supplement (Invitrogen). Cell cultures were terminated by fixation with cold 100% methanol for 10 min at 4°C and stored in phosphate-buffered saline (PBS) at 4°C until further processing.
Reverse Transcription-PCR (RT-PCR) Analysis of Skeletal Muscle Gene Expression
Total cellular RNA was extracted from 1–2 times; 105 cultured cells of passage number 5–7 using the Nucleospin II kit (Macherey-Nagel, Düren, Germany) and following the protocol provided by the manufacturer. The RNA was eluted in 60 μl of RNAse-free water and its concentration was determined with a NanoDrop ND-1000 (Isogen Life Science, De Meern, the Netherlands) spectrophotometer.
cDNA of each sample was generated from 2 μg of RNA, using 0.25 μg random hexamers and 25 nmol dNTPs (both from Invitrogen) in a total volume of 26.5 μl. This mixture was incubated for 5 min at 65°C to denature the RNA and subsequently placed on ice. After the addition of 10 μl of 5× First-Strand Buffer (Invitrogen), 5 μl of 100 mM dithiothreitol (Invitrogen), and 6 μl of water, the sample was incubated at 25°C for 5 min to facilitate primer annealing. Subsequently, 2.5 μl of SuperScript II (Invitrogen) was added and the mixture was incubated for 60 min at 42°C followed by 15 min at 70°C. The cDNA samples were then stored at −20°C.
Five microliters of 10-fold diluted cDNA was added to 15 μl PCR mixture containing: 1× PCR Taq buffer (Fermentas, St. Leon-Rot, Germany), 100 nmol MgCl2 (Fermentas), 4 pmol each of the forward and reverse primer (Invitrogen), 10 nmol dNTPs, and 1 U Taq polymerase (Fermentas). The PCRs were run in a PTC-240 Tetrad 2 DNA Engine Peltier thermal cycler (MJ Research, Ramsey, MN). The cycling conditions consisted of 1 min at 94°C followed by a variable number of cycles of 94°C for 30 s, 30 s at the annealing temperature of the primer pair used and 72°C for 30 s, and a final elongation step of 2 min at 72°C. Number of cycles and annealing temperature of each primer pair are listed in Table 2. As internal controls for the quantity and quality of the RNA samples, RT-PCRs targeting transcripts of the housekeeping genes glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin (ACTB) were performed in parallel. Commercially obtained total cellular RNA derived from skeletal muscle of human fetuses and adults (Agilent Technologies, Santa Clara, CA) was also subjected to RT-PCR analysis to provide positive controls. RT-PCRs of RNA samples from HeLa, HepG2, and VH10 cells were included to determine the specificity of the marker genes. PCRs carried out with water served as negative controls.
Primer Sequences and Conditions Used for RT-PCR

bp, base pairs; Ta, annealing temperature; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ACTB, β-actin; MYOD1, myogenic differentiation 1; MYOG, myogenin (myogenin factor 4); MYF5, myogenic factor 5; MYF6, myogenic factor 6 (herculin); MEF2C, myocyte-specific enhancer factor 2C; TNNI1, troponin I type 1 (skeletal, slow); TNNI2, troponin I type 2 (skeletal, fast); DES, desmin; MYH2, myosin, heavy chain 2, skeletal muscle, adult; DMD, dystrophin.
Immunocytology
MSCs (passage number 6) and myoblasts that were cultured on glass coverslips were processed for immunocytology as previously described (33) prior to their incubation with three different anti-human dystrophin mMAbs [NCL-DYS3 (Leica Biosystems Newcastle, Newcastle upon Tyne, UK: IgG2a), NCL-DYS2 (Leica Biosystems Newcastle: IgG1), or MANDRA1 (Development Studies Hybridoma Bank, University of Iowa, Iowa City, IA: clone 7A10; IgG1)] at a dilution of 1:5 in PBS-10 mM glycine (PBS-G)/5% FBS. Next, the cells were stored overnight at 4°C, washed thrice with PBS-G, and incubated with highly cross-adsorbed Alexa Fluor 568-conjugated goat anti-mouse IgG (H+L) antibodies (Invitrogen: 1:100) for 1 h at 4°C. Cells were then washed with PBS-G three more times for 3 min each. Finally, nuclei were stained with Hoechst 33342 (Invitrogen; 10 μl/ml in PBS) for 10 min at room temperature (RT). Cells were washed thoroughly with PBS-G prior to microscopic analysis.
Immunoblotting
Protein extracts were prepared from cultured cells of passage number 6 by adding lysis buffer consisting of 100 mM Tris-HCl (pH 6.8), 20% glycerol, and 25% of sodium dodecyl sulfate (SDS). The protein concentration in each cell lysate was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Etten-Leur, the Netherlands) according to the manufacturer's instructions. Subsequently, 0.4–1 μg of protein was diluted in 100 μl of 75 mM Tris-HCl (pH 6.8), 15% SDS, 5% β-mercaptoethanol, 20% glycerol, and 0.001% bromophenol blue. Proteins were separated in a discontinuous 4–7% SDS-polyacrylamide gel and wet-blotted to a 0.2-μm pore size nitrocellulose membrane (Whatman Protran BA83; GE Healthcare Europe, Diegem, Belgium). Blots were blocked with 5% nonfat milk powder (ELK; Campina, Woerden, the Netherlands) in 150 mM NaCl, 10 mM Tris-HCl, pH 8.0 (TBS) followed by an overnight incubation with the dystrophin-specific mMAb NCL-DYS1 (Leica Biosystems Newcastle: IgG2a; diluted 1:125 in TBS plus 0.05% Tween 20). IRDye 800CW-conjugated goat anti-mouse IgG (H+L) antibodies (LI-COR Biosciences, Lincoln, NE: 1:5,000) were used for detection. Visualization and quantification was done with the Odyssey infrared imaging system and accompanying software (both from Li-COR).
Heterotypic Cell Fusion Assay
The capacity of MSCs from the various human sources to fuse with the murine C2C12 myoblasts was assessed in a coculture system slightly different from that previously described (31). Twenty thousand MSCs of passage number 5–7 and C2C12 cells were plated alone (homotypic culture) or in a 1:1 ratio (10,000 cells of each type; heterotypic culture) in 24-well cell culture plates (CELLSTAR; Greiner Bio-One) in GM and cultured in humidified air with 5% CO2 until 90% confluency. GM was then replaced by myogenic differentiation medium. When large myotubes were observed (usually after 2–3 days), cells were fixed with cold 100% methanol for 10 min at 4°C, air dried, and stored in PBS at 4°C.
Before immunostaining, fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min at RT. Nonspecific binding sites were blocked by a 30-min incubation with PBS/5% FBS. The cells were then incubated overnight in a moisture chamber with mMAbs specific for human lamin A/C (Vector Laboratories, Burlingame, CA: clone 636; IgG2b; 1:200) and for fast-twitch skeletal myosin heavy chain (skMHC-II; Sigma-Aldrich: clone My-32; IgG1; 1:250). Next, the cells were incubated for 1 h with Alexa Fluor 568-conjugated goat anti-mouse IgG2b (Invitrogen) antibodies followed by a 1-h incubation with Alexa Fluor 488-conjugated goat anti-mouse IgG1 (Invitrogen) antibodies, both diluted 1:600 in PBS/5% FBS. Finally, nuclei were stained by a 10-min incubation with 10 μg/ml Hoechst 33342 in PBS. All incubations were performed at 4°C and each staining step was followed by three thorough washes with PBS. Microscopic analysis and capture of images were performed as described previously (33).
Lentiviral Vector Transduction
AT-, BM-, and SM-MSCs of passage number 4 of donor 2 were transduced with the vesicular stomatitis virus G protein-pseudotyped self-inactivating human immunodeficiency virus type 1 vector LV.C-EF1a.bGal (23). Twenty-four hours after seeding at a concentration of 4 times; 104 cells/cm2, MSCs were incubated for 4 h at 37°C with vector particles at a multiple of infection of 2 in GM supplemented with 8 μg/ml hexadimethrine bromide (Sigma-Aldrich) as previously described (23). Transduction efficiency was determined 14 days later by staining the cells with X-gal solution containing 2 mM 5-bromo-4-chloro-3-indolyl-β-D-galactosidase (X-gal; Sigma-Aldrich) as described previously (32). MSCs of all three sources were more than 90% β-gal-positive (β-gal+) at this time point as well as after multiple ex vivo cell doublings.
In Vivo Myoregeneration Assay
NOD/LtSz-scid/scid/J (NOD/SCID) mice were bred in the animal facilities of the LUMC from pairs originally purchased from Jackson Laboratories (Bar Harbor, ME). Male and female mice aged 8–12 weeks were used and housed as previously specified (48). All experiments were performed according to a study protocol approved by the animal ethics committee of the LUMC.
The in vivo assay used to assess the contribution of MSCs to muscle regeneration has been previously described (23). Briefly, 5 × 105 LacZ-transduced MSCs (derived from donor 2; amplified in GM containing 0.5 ng/ml FGF2 and used at passage number 8) were injected at one spot into the center of a TAM 24 h after injury induction by Naja mossambica mossambica CTX (Sigma-Aldrich). TAMs injected with CTX only were used as controls. Mice were sacrificed 30 days after cell injection. The TAMs were excised and either fixed in 4% phosphate-buffered formaldehyde or cryopreserved. Fixed TAMs were embedded in paraffin and stained with X-gal. Tissue sectioning and counting of β-gal+ myofibers along the entire TAM were performed as reported (23).
Immunohistology
Cryopreserved TAMs were cut into 6-μm-thick serial transversal cross sections. Eight consecutive sections were mounted on precoated Super Frost Plus slides (Menzel-Gläser, Braunschweig, Germany) and stored at −80°C.
First, sections on each 5th slide were stained with X-gal as previously described (23). This enabled the identification of areas along the TAMs containing hybrid myofibers. Slides adjacent to those containing β-gal+ myofibers were stained with antibodies specific for human proteins.
Tissue sections chosen for immunostaining with human β-spectrin- and human lamin A/C-specific mMAbs were fixed for 10 min with ice-cold acetone (Mallinckrodt Baker), air dried, and then incubated overnight at 4°C in a moist chamber with the anti-human β-spectrin antibody (Leica Biosystems Newcastle: clone RBC2/3D51; IgG2b; 1:20). Next, sections were incubated with Alexa Fluor 568-conjugated goat anti-mouse IgG2b antibodies (1:300) for at least 2 h at RT. Slides were then sequentially incubated for 1 h at RT with the lamin A/C-specific mMAb (1:200) and with the Alexa Fluor 568-conjugated goat anti-mouse IgG2b antibodies (1:500).
Tissue sections chosen for immunostaining with dystrophin-specific antibodies were thawed and (re)hydrated with PBS for 5 min at RT. Sections were then incubated with 1% IgG-free bovine serum albumin (Jackson ImmunoResearch, Suffolk, UK) for 30 min at RT. Next, the anti-human dystrophin antibody NCL-DYS3 was applied at a dilution of 1:5 followed by overnight incubation at 4°C. After washing with PBS, Alexa Fluor 568-conjugated goat anti-mouse IgG (H+L) antibodies (1:300) were added and sections were incubated for 2 h at RT.
In general every incubation step was followed by three 5-min washes with PBS. In all tissue sections nuclei were stained with Hoechst 33342 (10 μl/ml in PBS) by a 5-min incubation at RT. After washing with PBS, sections were mounted in Vectashield (Vector Laboratories) mounting medium. Light microscopic analysis and capture of images were performed as previously described (23).
Statistics
Results are expressed as mean ± SD. The data were analyzed by one-way analysis of variance. Differences between experimental groups (TAMs injected with MSCs from AT, BM, or SM) were evaluated by Student's t-test followed up by Bonferroni correction within groups. Statistical analysis was performed using GraphPad software. A value of p < 0.05 was considered significant. Pierce's criterion was applied for identifying outliers.
Results
Long-Term Growth Characteristics of MSCs From Different Human Tissues
The growth characteristics of AT-, BM-, and SM-derived human MSCs from three donors were compared in expansion cultures supplemented or not with FGF2 for up to 15 passages (>250 days). The data presented in Figure 1A show that addition of FGF2 to the culture medium caused an increase in the proliferation rate of the MSCs, which was independent of the cell source. The average PDT for each of the three types of MSCs from all donors about halved by the addition of FGF2.
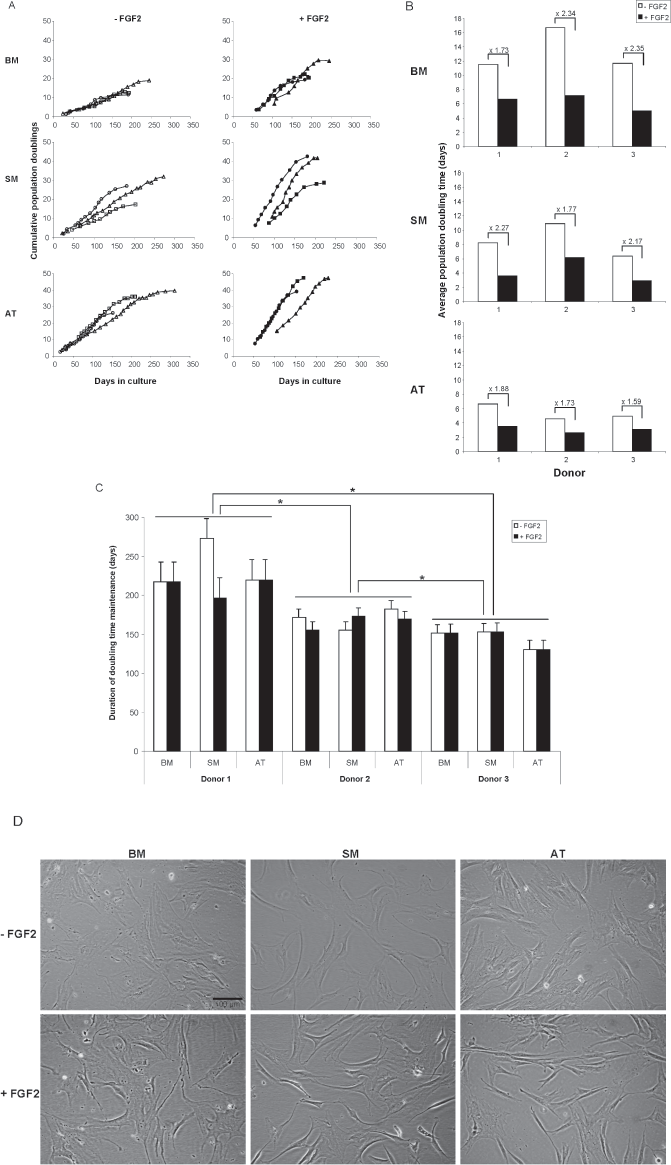
Long-term growth characteristics of mesenchymal stem cells (MSCs) derived from bone marrow (BM), synovial membrane (SM), and adipose tissue (AT) of three donors. (A) Cumulative population doublings (CPD) of cells that were cultured either with [+ fibroblast growth factor 2 (FGF2), closed symbols] or without FGF2 (– FGF2, open symbols). The data depicted by triangles, squares, and circles are derived from donor 1, 2, and 3, respectively. (B) Average population doubling time (PDT) of BM-, SM-, and AT-MSCs cultured with or without FGF2 (black and white bars, respectively) calculated from the linear part of the growth curves. Numbers above each pair of bars indicate the ratios of the average PDTs. Pooling all average PDTs per each tissue source the difference between AT- and BM-MSCs was statistically significant (p < 0.05). (C) Doubling time maintenance (DTM) of BM-, SM-, and AT-MSCs cultured with or without FGF2 (black and white bars, respectively). For all three donor pairs there was a statistically significant difference between the average DTM values (*p < 0.05). (D) Bright field images of parallel cultures of BM-, SM-, and AT-MSCs with (+ FGF2) or without FGF2 (– FGF2) supplementation revealed that cells had similar morphology. Original magnification: 10×.
AT-MSCs showed on average the highest number of CPDs (both in the presence and absence of FGF2) (Fig. 1A) and the shortest average PDT (Fig. 1B). The average CPDs of BM-, SM-, and AT-MSCs were 18.95 ± 6.51, 31.52 ± 9.51, and 39.32 ± 7.96, respectively, while the average PDTs were 9.77 ± 4.34, 6.36 ± 2.94, and 4.26 ± 1.47, respectively. Statistically significant differences (p < 0.05) for both parameters were only obtained between the AT-MSCs and the BM-MSCs.
A comparison of the DTM of all MSC samples (Fig. 1C) suggests that this characteristic is intrinsic to the donor and not related to the tissue of origin or the presence of FGF2. The average DTM values for cells of donor 1, 2, and 3 were 223.5 ± 25.8, 167.5 ± 10.65, and 144.7 ± 11.4 days, respectively. The differences between the values of the three donors were statistically significant (p < 0.05).
MSCs of the different tissues from all three donors did not display telomerase activity at both early and later passages irrespective of whether the cells were expanded in the presence or absence of FGF2 (data not shown). From all three donors parallel cultures of AT-, BM-, and SM-MSC with or without FGF2 supplementation revealed that cells had similar morphology (Fig. 1D).
Immunophenotype of MSCs From Different Human Tissues
The surface marker expression profile of the BM-MSCs (Fig. 2) was in general agreement with previously described results (16,18,63,81). The immunophenotype of the AT- and SM-MSCs was largely similar to that of the BM-MSCs apart from the surface expression levels of CD10, CD146, CD200, tissue nonspecific alkaline phosphatase (ALPL), and stage-specific embryonic antigen-4 (SSEA-4). CD10, CD146, and CD200 are expressed at moderate levels on the plasma membrane of AT-MSCs and at low levels or not at all on that of SM-MSCs and BM-MSCs. ALPL was detected at low levels only on the surface of BM-MSCs. The protein SSEA-4 is present on the plasma membrane of MSCs from all three sources, albeit at different levels with the highest and lowest surface levels of SSEA-4 occurring in SM- and BM-MSCs, respectively.
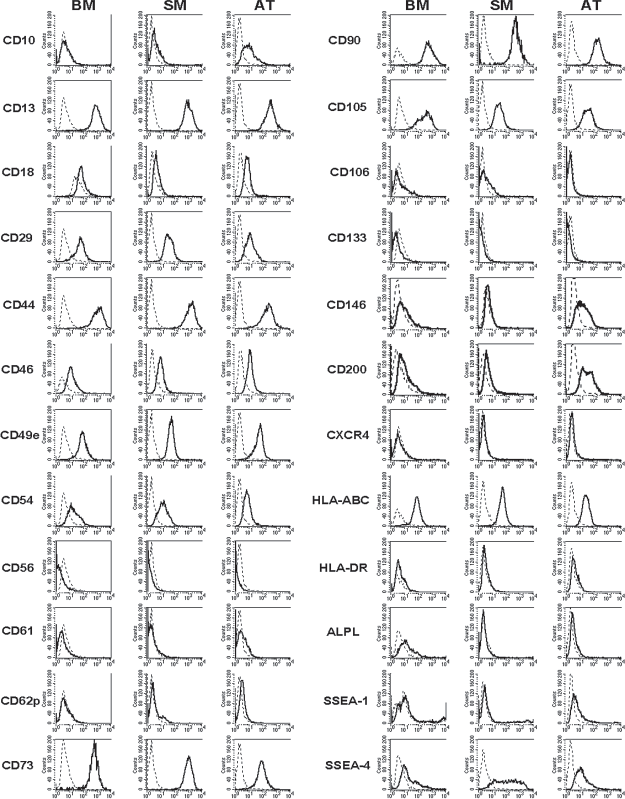
Immunophenotype of MSCs derived from different human sources. Presented are histograms of BM-, SM-, and AT-MSCs of donor 2. The surface antigen expression profile of BM-, SM-, and AT-MSCs of the other two donors was very similar. Solid lines correspond to the signals obtained with the specified mouse monoclonal antibodies (mMAbs); dotted lines show the signals of isotype-matched control antibodies.
Multilineage Differentiation of MSCs From Different Human Tissues
Culture-expanded MSCs derived from AT, BM, and SM of all donors were exposed to osteogenic, chondrogenic, or adipogenic differentiation conditions. In agreement with previous reports (62,88), the AT- and BM-MSCs readily differentiated into cells of all three lineages (Fig. 3). Remarkable was the limited osteogenic differentiation capacity of the SM-MSCs (Fig. 3h) of all three donors. Our finding is in line with that of Djouad et al. (26), who reported significantly less efficient osteogenic differentiation of SM-MSCs compared to that of BM-MSCs. However, the poor osteogenic differentiation of SM-MSCs in our experiments does not concur with the results published by De Bari et al. (20,22). This discrepancy probably reflects donor variability and/or dissimilarities in tissue harvesting as we cannot identify any other methodological differences between the two studies (ours and De Bari). Variation between different donors in osteogenic capacity of SM-MSCs isolated from the same site has been reported previously (20,45,71).

Differentiation of MSCs isolated from three different tissues. BM-, SM-, and AT-MSCs of donor 3 were cultured in adipogenic, chondrogenic, or osteogenic differentiation medium. (a, b, c) Oil red O staining for fat droplets. (d, e, f) Collagen type II labeling using a specific mMAb. (g, h, i) Alizarin red S staining for calcium deposits. Similar results were obtained with cells of the other two donors. Magnifications: 400× (a, b, c), 100× (c, d, e), 40× (g, h, i).
Expression of Skeletal Muscle Genes in Culture-Expanded Human MSCs
Total cellular RNA from culture-expanded MSCs was subjected to RT-PCR analyses to investigate the expression of skeletal muscle genes. These analyses included the myogenic regulatory factor (MRF) genes myogenic differentiation 1 (MYOD1; also known as myogenic factor 3; MYF3), MYOG (also known as MYF4 or MYOGENIN), MYF5, and MYF6 (also known as MRF4 or HERCULIN) and the myocyte-specific enhancer factor 2C (MEF2C) gene. Also studied were genes encoding proteins occurring later in the process of skeletal muscle differentiation like slow-twitch skeletal troponin I (encoded by the TNNI1 gene), fast-twitch skeletal troponin I (specified by the TNNI2 gene), desmin (encoded by the DES gene), skeletal myosin heavy chain IIa (specified by the MYH2 gene), and dystrophin (encoded by the DMD gene). As to the latter gene, primers were chosen in exon 1 and 3 of the mRNA to amplify only the striated muscle-specific splice variant of the DMD gene encoding the Dp427m isoform of dystrophin (hereinafter referred to as DMD transcript). The specificity of these primers was confirmed by the presence of amplification products of the correct size in adult and fetal skeletal muscle tissue samples.
Of the four MRF genes analyzed, PCR products of MYOG were detected in three samples (AT- and SM-MSCs of donor 2 and AT-MSCs of donor 3) (Fig. 4). PCR products of MEF2C were present in all MSC samples at similar levels as well as in HeLa and VH10 cells confirming the widespread expression of the MEF2C gene (81).
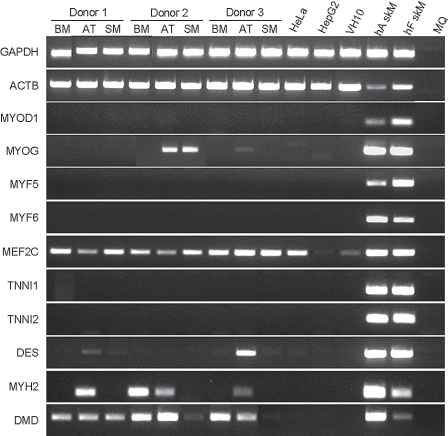
Myogenic gene expression profile of MSCs expanded in growth medium (GM). RT-PCR analysis of skeletal muscle marker gene expression in MSCs derived from BM, SM, and AT of three donors. Total RNA extracted from MSCs and from control cells/tissues [i.e., Hela, HepG2, and VH10 cells, human adult (hA skM) and fetal (hF skM) skeletal muscle] was subjected to RT-PCR with primers specific for the indicated genes (Table 2). MQ, water.
Transcription of the DES gene was evident in AT-MSCs of 2 donors (1 and 3) and that of the MYH2 gene in AT-MSCs of all donors and in BM-MSCs of donor 2. Unexpected was the detection of DMD transcripts in all MSC samples analyzed. In spite of the presence of dystrophin-encoding mRNA in the MSCs, the corresponding protein was not detected in these cells by either immunocytology or Western blotting (Fig. 5).

Culture-expanded MSCs do not contain detectable amounts of dystrophin. (A) Bright field and immunofluorescence images of BM-MSCs in GM (a–d) and of myotubes derived from LHCN-M2 myoblasts (MTWT; e, f) stained with a human dystrophin-specific mMAb and Alexa Fluor 568-conjugated goat anti-mouse IgG (H+L) antibodies (red; b, f) or with the secondary antibody only (d). Nuclei were stained with Hoechst 33342 (blue; b, d, f). Magnifications: 100× (a–d) and 200× (e, f). (B) Western blot analysis of dystrophin in BM-MSCs expanded in GM, HepG2 cells, iDMD myoblasts (MBDMD), myotubes derived from iDMD myoblasts (MTDMD), LHCN-M2 myoblasts (MBWT), myotubes derived from LHCN-M2 myoblasts (MTWT), and murine skeletal muscle (m skM).
Collectively, our PCR results demonstrate transcription of different skeletal muscle genes with the largest variety of transcripts occurring in AT-MSCs. The inconsistent occurrence of certain transcripts in cells of different donors may argue for their presence at very low levels.
MSCs From all Three Human Sources Form Hybrid Myotubes in Cocultures with Murine Myoblasts
In a previous study, we showed that human BM-MSCs contribute to myotube formation in cocultures with human myoblasts through cell fusion (31). Formation of hybrid myotubes by heterotypic cell fusion has also been observed in cocultures of human BM-MSCs and the murine C2C12 myoblasts (75). To get an impression of the number of human cells incorporated into hybrid myotubes for each of the MSC sources we employed the MSC-C2C12 coculture system. The number of human cells incorporated in individual myotubes was estimated with the aid of an antibody specific for human lamin A/C. Lamin A and C are nuclear proteins with a very low turnover rate (8), which allowed us to distinguish unambiguously between human and murine nuclei in skMHC-II-stained myotubes. Figure 6 shows examples of multinucleated syncytia containing one to four human nuclei in cocultures of C2C12 cells and human MSCs from each of the three sources. The number of hybrid myotubes was low in all cocultures examined. On average three, six, and seven of such myotubes were observed in mixed cultures containing AT-, BM-, or SM-MSCs, respectively (n = 3). Moreover, no conspicuous differences were observed in the frequency of hybrid myotubes or in the average frequency of human nuclei per hybrid myotube (data not shown). Finally, exposure of monocultures of MSCs from all three sources to myogenic differentiation medium (negative control) did not show homotypic cell fusion or any other morphological signs of skeletal myogenesis (data not shown).
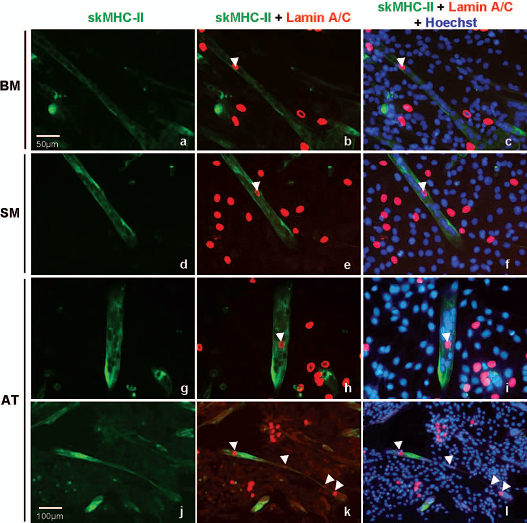
Hybrid myotubes derived from fusion of human MSCs and murine myoblasts. Human BM-, SM-, and AT-MSCs of donor 3 were cocultured with C2C12 cells. The cultures were fixed and stained with mMAbs specific for skMHC-II (green; sarcomeres), human lamin A/C (red; nuclear membrane) and Hoechst 33342 (blue; nuclei). Similar results were obtained with cells of the other two donors. Arrowheads indicate human nuclei inside hybrid myotubes. Notice in (k) and (l) the presence of four human nuclei in a single hybrid myotube. Magnifications: 200× (a–i), 100× (j–l).
The Contribution of MSCs From Three Different Human Sources to Skeletal Muscle Regeneration In Vivo
The participation of human AT-, BM-, and SM-MSCs in skeletal muscle regeneration was studied in immunodeficient mice with CTX-injured TAMs using LacZ-transduced cells from a single donor (donor 2).
As very large numbers of cells are required for this type of experiment, we choose to propagate the MSCs in medium supplemented with 0.5 ng/ml FGF2. Before starting this experiment, we first compared the frequency of hybrid myofibers in whole CTX-injured TAMs of NOD/SCID mice that received BM-MSCs expanded in the absence of FGF2 to that of animals injected with cells expanded in the presence of FGF2. Screening of 200 X-gal-stained transversal sections of each TAM at 30 days posttransplantation revealed an average of 27.7 ± 19.3 (n = 3) and 27.0 ± 4.2 (n = 2) β-gal+ hybrid myofibers in muscles injected with cells cultured without and with FGF2, respectively. Although the number of mice in each group was low, the very similar results obtained under both conditions let us to conclude that the presence of FGF2 during expansion cultures of MSCs does not affect their contribution to myoregeneration.
A similar histological analysis of TAMs transplanted with MSCs of the different sources revealed spread of single human cells throughout the entire muscle (data not shown) as observed previously with human BM-MSCs (23). The total number of hybrid (i.e., β-gal+) myofibers per treated TAM (Fig. 7) showed a large intragroup variation for mice transplanted with AT- or SM-MSCs. However, none of the values in these TAMs met the Pierce criteria for outliers. Even so, statistical analysis revealed that the average number of β-gal+ myofibers for AT-MSCs was significantly higher than for BM-MSCs (17.0 ± 14.5 vs. 4.6 ± 4.6, p < 0.05). The contribution of SM-MSCs to myofiber formation (10.0 ± 9.7) did not significantly differ from that of AT- or BM-MSCs.

Contribution of MSCs from different human sources to skeletal muscle regeneration in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice. (A) Number of hybrid (i.e., β-gal+) myofibers per individual tibialis anterior muscle (TAM) at 30 days after implantation of LacZ-transduced BM-, SM-, and AT-MSCs. Eight TAMs per tissue source were analyzed. The difference in the number of hybrid myofibers between AT-MSCs and BM-MSCs is statistically significant (*p < 0.05). (B) Paraffin sections of TAMs collected 30 days after local injection of BM-, SM-, and AT-MSCs were stained with X-gal and counterstained with hematoxylin, phloxin, and saffron. Magnifications: 40× (a–c), 200× (d–f).
In cryosections of TAMs transplanted with cells of any of the three sources we could identify myofibers expressing human β-spectrin or human dystrophin (Fig. 8). In general, the frequency of myofibers positive for human β-spectrin was higher than that of those containing human dystrophin. Furthermore, only few myofibers stained positive for both human dystrophin and human β-spectrin. These proteins were not present in the human lamin A/C-positive mononucleated cells in the muscle interstitium. The analysis of consecutive sections revealed the presence of some myofibers that stained positive for human β-spectrin, human dystrophin, and β-gal (Fig. 8C).

Immunodetection of human skeletal muscle proteins in regenerating murine TAMs following local injection of human MSCs. (A) Transversal tissue sections of CTX-treated TAMs collected 30 days after implantation of LacZ-transduced MSCs derived from BM (a, d) SM (b, e), or AT (c, f) and stained with antibodies specific for human β-spectrin (lining the sarcolemma; red) and lamin A/C (nuclear membrane; red; a–c) or with a mMAb directed against human dystrophin (lining the sarcolemma; red; d–f). All sections were costained with Hoechst 33342 (blue; nuclei). Magnifications: 1,000× (a, b), 630× (c–f). (B) Transversal tissue sections of control samples (i.e., human adult skeletal muscle) (hA skM; a, d), uninjured murine TAM (uninjured TAM; b, e), and murine TAM excised 31 days after CTX injection (CTX-treated TAM; c, f) stained as in (A). Magnification: 400×. (C) Consecutive transversal cross sections of two different TAMs (upper and lower row) treated with CTX and collected 30 days after implantation of LacZ-tagged AT-MSCs. The symbols in the photographs #, *, and ∘ mark the same three myofibers stained in different sections as follows: in (a) and (d) showed sections stained with antibodies specific for human β-spectrin (lining the sarcolemma; red) and human lamin A/C (nuclear membrane; red); sections in (b) and (e) were stained with an antibody specific for human dystrophin (lining the sarcolemma; red). The sections in the left and middle panel (a, b, d, e) were costained with Hoechst 33342 (nuclei; blue). The sections in the right panel (c, f) were stained with X-gal (cytoplasm, blue) and nuclear fast red (NFR; nuclei; red). Magnifications: 630× (a, b, d, e), 400× (c, f).
Discussion
MSCs are regarded as a platform for cell- and gene-based therapies that hold promise for clinical application in regenerative medicine, immune modulation, and cancer treatment. After encouraging results were reported with BM-derived MSCs in a variety of conditions (9,50,52,73), the use of MSCs from alternative sources became of practical interest. A comparison of the inherent properties and the in vivo therapeutic potential of human MSCs from different tissues of the same donor is not yet available. In the current study, the properties of MSCs of AT, BM, and SM from three human donors were compared both in vitro and in vivo. By analyzing different MSC sources of the same donors, donor-related factors as cause of potential differences in the performance of MSCs from different tissues could be excluded. The parameters evaluated were selected according to their significance for the applicability of the cells in particular for the treatment of myopathies.
Ex Vivo Growth Characteristics of MSCs From Different Human Tissues
Long-term culture of human AT-, BM-, and SM-MSCs from three donors revealed clear differences in their ex vivo growth characteristics. Our results signify that of these three different types of MSCs, AT-MSCs possess the highest replication potential. This finding differs from that of Sakaguchi et al. (71), who reported that the replication rate of AT-MSCs is much lower than that of BM- and SM-MSCs. A noticeable difference between the two studies is the age of tissue donors: 69 ± 3.51 years in our study versus 23 ± 6 years in the study performed by Sakaguchi and colleagues (71).
So far there is no consensus as to the influence of organismal age on cell replication in vitro (13,36,38,43,55). The few studies addressing the effect of donor age on the in vitro replication of human MSCs have led to conflicting conclusions. De Bari et al. (20), using MSCs isolated from SM, found no difference in replication rate for a period of more than 100 days between cells from young and elderly donors. In contrast, a negative relation between donor age and replication capacity was reported for BM-MSCs by Stenderup et al. (79). Moreover, the replication rate reported by Stenderup and colleagues (79) for BM-MSCs of elderly donors cultured in the absence of FGF2 closely resembled that found by us in the present study. Unfortunately, the relation between donor age and proliferation of AT-MSCs to our knowledge has not been studied. One explanation for the discrepancy between our results and those of Sakaguchi et al. (71) is that BM-MSCs undergo a much faster decline in replication rate with donor age than AT-MSCs. This may result in a reversal of the ranking of the proliferation rate of MSCs from the two sources in the elderly. If so, ageing of MSCs, expressed as changes in their proliferation properties, may occur in different tissues at different rates.
Our data further showed a strong effect of FGF2 supplementation on the replication of human MSCs from all three sources examined (Fig. 1), as manifested by a reduction of their average PDT. Interestingly, the DTM of the cells was not affected by FGF2 but seems to be determined by the donor. The fact that our donors were of similar age together with the absence of telomerase activity in MSCs [(2,3,87), this article] and the previously reported relationship between initial telomere length and MSC proliferation capacity in culture (76) suggest that differences in telomere length between the starting materials determine DTM.
In addition, the absence of telomerase activity in MSCs from the three human sources and their finite ex vivo replication capacity argue against the immortalization or transformation of these cells during long-term culture.
MSC Immunophenotype and Multipotency
Although culture-expanded human MSCs are assumed to represent a heterogeneous population (5,45,64,68), immunophenotypic characterization of the MSCs from AT, BM, and SM revealed, in accordance with previous reports (34,65,71), that more than 95% of the cells in each culture type express the well-defined MSC-associated surface markers CD44, CD73, CD90, and CD105. Cells from the different sources and donors strongly resembled each other in surface antigen expression profile except for differences in the surface levels of CD10, CD146, CD200, ALPL, and SSEA-4 (Fig. 2), with the first three glycoproteins being expressed at higher levels on the plasma membrane of AT-MSCs.
CD10 is a type II integral membrane glycoprotein with neutral endopeptidase activity, present on the surface of many mammalian cell types (e.g. kidney, brain, cells of the immune system). Various research groups reported the presence of CD10 on the surface of human adult MSCs of different tissues. Although there is no consensus as to the plasma membrane levels of this marker and its function, the available data suggest that the frequency of CD10+ cells is higher in human AT-MSC cultures than in ex vivo propagated SM-MSC populations (57,71).
CD146, a heavily glycosylated type I transmembrane protein, distinguishes within the BM mononuclear cell fraction between hematopoietic progenitors (CD146–) and cells associated with the subendothelial stroma (CD146+) (70). The CD146+ cells of BM have the general accepted characteristics of MSCs (i.e., self-renewal capacity, a distinctive cell surface antigen profile, and multilineage differentiation capacity) (78). The use of CD146 as a general marker for MSCs has been disputed due to differences in its surface expression between MSCs derived from different donors and/or sources (44,61). The latter finding was confirmed in our study, which showed that the surface of culture-expanded AT-MSCs contains a much higher number of CD146 molecules than that of culture-amplified BM- or SM-MSCs.
CD200 is a type I integral membrane glycoprotein expressed on the plasma membrane of a wide variety of cell types. In freshly isolated BM cell preparations, the CD200+ cell fraction was highly enriched for cells with MSC properties (24). The only available data on cultured MSCs is from BM-derived cells, which were shown to down regulate this marker upon culture (5). The latter finding fits our observation that culture-expanded BM-MSCs as well as SM-MSCs displayed very few molecules on their surface. AT-MSCs, on the other hand, showed high CD200+ surface expression after ex vivo propagation. Considering the proposed contribution of CD200 to the immunosuppressive properties of other cell types (11,80,83), our findings provide a rationale for investigating whether a correlation exists between the immunosuppressive ability of MSCs from different sources and their CD200 surface level.
MSCs isolated from various adult human tissues (30,65,67,77) have also been shown to contain several embryonic stem cell markers including SSEA-4, octamer-binding transcription factor 4 (OCT4), sex determining region Y-box 2 (SOX2), NANOG, and ALPL (an ectoenzyme attached to the plasma membrane via a glycophosphatidylinositol anchor). Our finding that AT- and BM-MSCs express low levels of ALPL and intermediate/low levels of SSEA-4 at their surface corroborates with previous results (65). In contrast, SM-MSCs did not contain detectable amounts of ALPL at their plasma membrane and were heterogeneous in SSEA surface level, which ranged from very low to very high. The significance of the latter finding is not clear. Nevertheless, the absence of a direct relation between ALPL and SSEA-4 surface levels and MSC replication rate and/or multipotency (compare AT-MSCs to SM-MSCs) raises doubt as to whether these proteins have similar functions in adult cells as in embryonic cells.
Myogenic Characteristics of MSCs From Different Human Tissues and Their Contribution to Skeletal Muscle Regeneration
The myogenic propensity of human MSCs of the three sources has been individually investigated by others on the basis of myogenic gene expression profile and fusion capacity (15,22,25,54,72,88). Single-cell microarray analysis of cultured BM-MSCs showed that they express genes characteristic of many different cell types, including myocytes (74). However, using RT-PCR analysis of total cellular RNA extracted from culture-expanded monoclonal and polyclonal BM-MSC populations, Delorme and colleagues (24) could not demonstrate activation of skeletal muscle-specific transcription factor genes suggesting that BM-MSCs are not primed to the myogenic lineage. Our RT-PCR results with BM-MSCs (Fig. 4) fully agree with those of Delorme et al. (24). However, in some AT- and SM-MSC samples we did detect expression of the MYOG gene (Fig. 4). Interesting was the detection of mRNA sequences of the genes encoding desmin, skeletal myosin heavy chain IIa and dystrophin, in samples of culture-expanded MSCs (Fig. 4). Thus far, the presence of desmin and MYH2 transcripts in MSCs has only been demonstrated for cells derived from fetal BM (7). The latter cells were found to readily undergo skeletal muscle cell differentiation in culture following galectin-1 treatment, which underscores their myogenic potential and distinguished them from adult BM-MSCs. The presence of DMD transcripts in culture-amplified adult human MSCs has not been reported before. Our failure to detect dystrophin in human MSCs (see Fig. 5) and our observation that these cells do not show homotypic cell fusion or any other morphological signs of skeletal myogenesis after exposure to myogenic differentiation medium or when cocultured with C2C12 cells suggests that in MSCs DMD mRNA may have a nonprotein-coding function.
In coculture with murine C2C12 myoblasts, MSCs of all three human sources displayed a similar myogenic fusion activity, contributing at a low frequency to hybrid myotube formation (Fig. 6).
The myogenic potential of MSCs of all three sources was also demonstrated in vivo in TAMs recovering from CTX-induced injury. However, the extent at which MSCs of the different human sources contributed to myofiber formation/regeneration differed. Since murine TAMs contain approximately 2,000 skeletal muscle cells (23), the percentage of myofibers with a contribution of human cells at 30 days after cell transplantation was 0.85%, 0.5%, and 0.23% for the muscles injected with AT-, SM-, and BM-MSCs, respectively. The number of hybrid myofibers in TAMs transplanted with AT-MSCs was significantly higher than that in TAMs transplanted with BM-MSCs. The human versions of β-spectrin and dystrophin, two proteins that are synthesized at a relatively late stage of skeletal muscle differentiation, were detected in hybrid myofibers (Fig. 8) but not in the human lamin A/C-positive mononucleated cells. The latter observation, which is consistent with the finding that the cultured MSCs contained neither dystrophin (Fig. 5) nor β-spectrin (data not shown), implies that nuclei of human MSCs of all three sources can undergo myogenic reprogramming in the regenerating murine skeletal muscle environment. Interestingly, the frequency of myofibers positive for human β-spectrin was higher than that of myofibers containing human dystrophin and the two human skeletal muscle proteins were co-detected in only a fraction of the hybrid myofibers. Similar observations were made for murine muscles implanted with human myoblasts (10) and may be associated with myofiber maturation. While β-spectrin is made at an early stage of myofiber maturation, the presence of dystrophin marks the end of this process.
In summary, our study reveals the existence of differences between MSCs derived from different human tissues. Most prominent were the higher proliferation rate and expansion capacity as well as the greater contribution to in vivo myogenesis of AT-MSCs compared to those of SM- and BM-MSCs. Moreover, AT is abundantly present and easily accessible and contains a relatively high frequency of MSCs. This makes AT the tissue of preference for the acquisition of MSCs to be used for the further development of MSC-based treatments of myopathies.
Footnotes
Acknowledgments
AT, BM, and SM samples for the isolation of MSCs were kindly provided by S. Hogendoorn and R. G. H. H. Nelissen, Department of Orthopaedic Surgery, LUMC. This study was supported by a scholarship to A. S. de la Garza-Rodea from the Universidad Autónoma de Nuevo León, Monterrey, Mexico and by research grants from the Association Française contre les Myopathies (AFM; 12259-SR-GROUPE E) and the Duchenne Parent Project. The authors declare no conflicts of interest.