
Abstract
Endothelial cells (ECs) are involved in the process of angiogenesis, the outgrowth of new vessels from preexisting blood vessels. If available in sufficiently large numbers, ECs could be used therapeutically to establish blood flow through in vitro engineered tissues and tissues suffering from severe ischemia. Adipose tissue (AT) is an easily available source of large number of autologous ECs. Here we describe the isolation, in vitro expansion, and characterization of human AT derived ECs (AT-ECs). AT-ECs proliferated rapidly through 15–20 population doublings. The cultured cells showed cobblestone morphology and expressed EC markers including CD31, CD144, eNOS, CD309, CD105, von Willebrand factor, CD146, CD54, and CD102. They bound Ulex europaeus agglutinin I lectin and took up DiI-Ac-LDL. The AT-ECs formed capillary-like tubes in Matrigel in vitro and formed functional blood vessels in Matrigel following subcutaneous injection into immunodeficient mice. In conclusion, AT-ECs reach clinically significant cell numbers after few population doublings and are easily accessible from autologous AT, which also contains mesenchymal stem cells/pericytes. Thus, AT yields two cell populations that may be used together in the treatment of tissue ischemia and in clinical applications of tissue engineering.
Introduction
Therapeutic strategies based on the delivery of angiogenic cells have been attempted for peripherovascular disease (40,46) and ischemic heart disease (9), so far with modest functional improvement. Generation of new blood vessels is also a major issue in tissue engineering (20,32,39). Here different approaches have been used to overcome the problem of vascularization, such as incorporation of angiogenic factors into the engineered tissues to promote the ingrowth of microvessels, prevascularization of matrices prior to cell seeding, and seeding of endothelial cells (ECs) along with other cell types (27). However, to date no cell-based therapeutic strategy has been able to robustly generate new blood vessels in engineered or ischemic tissues.
The most popular candidates for strategies of cell-based therapeutic neovascularization are the endothelial progenitor cells (EPCs) (14,32). EPCs were first isolated from human peripheral blood by Asahara et al. (2). Two distinct EPC populations are observed with different growth characteristics, which are referred to early and late-outgrowth EPCs (31). Recently, the vasculogenic potential of EPCs from adult peripheral blood and human cord blood was described and compared (24). EPCs from cord blood were found to be superior to adult cells both in proliferative potential and vasculogenesis, but the vasculogenic capacity of both populations deteriorated rapidly with time in cell culture. In another study, endothelial colony-forming cells were isolated from unmanipulated human peripheral blood (34). These cells are easily available, but because the precursor frequency is low, prolonged in vitro cell expansion will be required in order to get an appropriate number of cells for therapy.
Mature ECs can also be candidates for cell therapy applications. These cells are mostly derived from the umbilical vein or the aortic endothelium. Both these populations may be expanded in vitro, although they do not normally proliferate over many passages (17). However, in terms of human cell therapy, the main problem associated with the use of these cells is that they will always be allogeneic to the patient, and therefore likely to be rejected by immunological mechanisms. Microvascular ECs (MVECs) resemble late-outgrowth EPCs (3). As such, they should be promising cells for therapeutic neovascularization. However, MVECs are most commonly obtained from sources such as newborn foreskin, the retina, and the myometrium, all sources that in practical terms will be allogeneic to the patient.
Adipose tissue (AT) is an easily available source of large number of autologous cells for cell therapy applications. Mesenchymal stem cells (MSCs) from AT have been isolated and characterized (4,49), and are currently being tested in clinical trials (12). MSCs are frequently obtained from the stromal-vascular fraction (SVF) of AT cells. The SVF of AT cells has also been shown to contain cells with angiogenic potential (30,33). Several attempts have been made to isolate ECs from SVF using differential plastic attachment and positive selection strategies (1,10,18,19,37). However, there is still a need for data on the phenotype of the AT-EC population, and how the phenotype and gene expression of this cell population is modulated by in vitro culture. These data, combined with experiments showing that the cells are able to form vessels in vivo, are required before AT-ECs may be accepted for clinical trials of EC transplantation.
Here we describe the isolation, purification, expansion, and extensive characterization, both at the uncultured and cultured stage, of the EC population obtained from the SVF from AT. Based on our previous observation that most of the AT-ECs did not express CD44, while the majority of AT-MSCs did express this marker (4), we chose to purify the AT-ECs from the SVF by removing CD44+ cells. We demonstrate that AT-ECs have endothelial morphology and phenotype, that they form capillary-like tubes in Matrigel in vitro and functional blood vessels in vivo. One important aspect of these cells, which separates them from other ECs, is that they derive from a source that is easily accessible in all patients. They can be obtained in very large numbers, which means that clinically relevant numbers of cells can be obtained after only a few population doublings in vitro. Importantly, they derive from a tissue that also yields MSCs (4,49,50). Recent studies on MSC populations suggest that these derive from blood vessel walls and that they may in fact be identical to the pericytes (5–7). Small blood vessels are composed of ECs surrounded by basal lamina and loosely covered by pericytes, which are embedded in the basement membrane of the microvessels underlying the ECs (23,38,41). The pericytes play a role in vascular stabilization by interaction with ECs (42) and can stimulate angiogenesis (26,45). Thus, AT may provide two cell populations required for vasculogenesis.
Materials and Methods
All reagents were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise stated.
Isolation of AT-ECs
AT was obtained from liposuction material from abdominal regions of three healthy female donors (age 24–40 years) undergoing cosmetic surgery. The donors provided informed consent, and the collection and storage of AT, AT-ECs, and AT-MSCs were approved by the regional committee for ethics in medical research. The SVF was separated from AT as described previously (4). Briefly, lipoaspirate (300–600 ml) was washed repeatedly to remove erythrocytes and leukocytes with Hanks' balanced salt solution (HBSS; Gibco, Invitrogen, Paisley, UK) containing antibiotics (100 IU/ml penicillin and 100 IU/ml streptomycin) and 2.5 μg/ml amphotericin B. This material was then digested for 75 min using 0.1% collagenase A type 1 at 37°C, applying constant gentle rotation. Following centrifugation at 400 × g for 10 min at room temperature the pellet was resuspended in HBSS containing antibiotics and 2% fetal bovine serum (FBS; BioWhittaker, Lonza, Verviers, Belgium), and the cells were filtered through 100-μm and then 40-μm cell sieves (Becton Dickinson, San Jose, CA). SVF cells were obtained from the interface after Lymphoprep gradient separation (Axis Shield, Oslo, Norway) at 400 × g for 30 min, washed, and resuspended in HBSS containing antibiotics and 2% FBS. Cell counts and viability assessment were performed using acridine orange/ethidium bromide staining and a fluorescence microscope (Eclipse E600, Nikon, Kanagawa, Japan).
To obtain AT-ECs from the SVF, CD44+ cells were removed using Dynabeads (Dynabeads Pan Mouse IgG, Invitrogen Dynal AS, Oslo, Norway) according to the manufacturer's description. Briefly, Dynabeads were washed, then precoated with monoclonal anti-CD44 antibody (Caltag Laboratories, Invitrogen, Carlsbad, CA) at the concentration of 0.6 μg antibody per 25 μl Dynabeads and incubated for 2 h with gentle tilting and rotation. Dynabeads were then washed again and approximately 400 × 106 magnetic CD44 beads were added to 100 × 106 SVF.
In Vitro Expansion of AT-ECs
AT-ECs were plated at 2 × 106 cells per 75-cm2 tissue culture flask (Nunc, Roskilde, Denmark) coated with 1% gelatin. Cells were maintained at 37°C in an atmosphere of 5% CO2 in humid air using EC growth medium (MCDB 131; Gibco) supplemented with 7.5% FBS, 1% L-glutamine, 1 ng/ml basic fibroblast growth factor, 1 μg/ml hydrocortisone, 10 ng/ml epidermal growth factor, 50 μg/ml gentamicin, and 250 ng/ml amphotericin B. Cells still in suspension were removed on day 4–5 and the medium was replaced. When the cells reached 80–90% confluency, cell adherence was interrupted by trypsin-ethylenediaminetetraacetic acid (EDTA) solution for ECs and the cells were inoculated into new flasks at 10,000 cells/cm2. Additional purging of contaminating cells was performed using anti-CD44-coated Dynabeads if required at the first passage. Cells were routinely passaged every 3–4 days. Viable cells were counted at each passage. Growth curves were generated using GraphPad Prism version 5 software (GraphPad Software Inc., La Jolla, CA).
Isolation and In Vitro Expansion of AT-MSCs
To obtain AT-MSCs/pericytes for the Matrigel vascularization studies the CD44+ cells positively selected from the SVF of the same donors were cultured in Dulbecco's modified Eagle's medium/F12 (Gibco) containing 10% FBS and antibiotics. To remove remaining CD45+ leukocytes, at P2, magnetic beads directly coupled to mouse anti-human CD45 monoclonal antibodies (Caltag Laboratories), as described above, were mixed with the CD44-depleted cells at a ratio of 4 beads/cell. After 2 h of incubation the CD45+ cells attached to beads were removed using a magnet, and the remaining cells were placed back in cell culture for further in vitro expansion.
Characterization of AT-ECs
The isolated AT-ECs from all the three donors were characterized at the uncultured stage (P0), at early (P2), middle (P6), and late (P12) passages. The endothelial nature of the cells was confirmed by real-time reverse transcription-polymerase chain reaction (RT-PCR), flow cytometry, Ulex europaeus agglutinin I lectin (UEA-1) binding, and DiI complex acetylated low-density lipoprotein (DiI-Ac-LDL) uptake as well as in vitro and in vivo angiogenesis assays.
Real-Time RT-PCR Analysis
Total RNA was extracted from cells at different passages using RNAqueous-Micro Kit (Applied Biosystems/Ambion, Austin, TX) according to the manufacturer's protocol and treated with DNase I (Applied Biosystems/Ambion). Reverse transcription (RT) was performed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) following the manufacturer's protocol with 200 ng total RNA/20 μl RT reaction. Relative quantification was performed using the 7300 Real-time RT-PCR system (Applied Biosystems) and TaqMan Gene Expression Assay (Applied Biosystems) reagents according to the manufacturer's description. The thermocycler parameters were 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. All primers are provided in Table 1. All samples were run in triplicate. After verifying the stable expression of CDKN1A, this gene was selected as endogenous control. The mRNA levels were normalized to the expression of CDKN1A for each sample. Relative mRNA levels were calculated by the comparative CT method, where the threshold cycle (CT) values are given by the 2–ΔΔCt formula (22). Differential expression of all markers was calculated in relation to the corresponding uncultured cells. Graphs were created with GraphPad Prism version 5 software. As a positive control, primary adult human dermal MVECs (P6) were purchased from PromoCell GmbH (Heidelberg, Germany). Human AT-MSCs and chondrocytes were used as negative controls for the endothelial phenotype.
List of Primers Used in Real-Time RT-PCR

Flow Cytometry
Flow cytometry was performed for determination of cell surface antigen expression of freshly isolated (uncultured) and cultured (P2, P6, and P12) AT-ECs from three different donors and of human dermal MVECs (P6). All monoclonal antibodies are provided in Table 2. Irrelevant control antibodies were included for each of the different fluorochromes. Cells were incubated with directly conjugated antibodies for 10 min, washed with phosphate-buffered saline (PBS) containing 1% FBS, and fixed in 1% paraformaldehyde (PFA). Cells were analyzed using a FACSCalibur flow cytometer applying BD CellQuest Pro Software (BD Biosciences).
List of Antibodies Used in Flow Cytometry

ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; HLA, human lymphocyte antigen; UEA-1, Ulex europaeus agglutinin 1.
Intracellular Staining for von Willebrand Factor
Flow cytometry was performed for determination of intracellular expression of von Willebrand factor (vWF) in freshly isolated (uncultured) and cultured (P2, P6, and P12) AT-ECs from three different donors. For intracellular staining, a BD Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences) was used according to the manufacturer's description. Briefly, cells were trypsinized, washed, centrifuged, and then resuspended in BD Cytofix/Cytoperm buffer. After 20-min incubation cells were washed in 1× BD Perm/Wash buffer and incubated for 30 min on ice with mouse anti-human vWF (Immunotech, Marseille, France). Cells were washed once again, incubated for 30 min on ice with goat anti-mouse Ig (H+L)-R-Phycoerythrin (RPE) (Southern Biotech), washed, and finally fixed with 1% PFA and analyzed by a FACSCalibur flow cytometer applying BD CellQuest Pro Software. As a negative control secondary antibody alone was used to assess nonspecific fluorescence.
DiI-Ac-LDL Uptake
To assess the uptake of DiI-Ac-LDL (Molecular Probes, Invitrogen, Eugene, OR), AT-ECs and AT-MSCs were seeded on gelatin-coated chamber slides (BD Falcon 4.0 cm2/chamber, BD Biosciences). After reaching confluence, cells were washed once with medium then incubated in medium containing 15 μg/ml of DiI-Ac-LDL for 4 h in a culture incubator at 37°C with 5% CO2. Then the cultures were washed with PBS and cells were fixed in 4% PFA. After fixation cells were washed again. Human chondrocytes were used as a negative control. All immunocytochemistry and immunohistochemistry slides were mounted with Vectashield mounting medium for fluorescence containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories Inc., Burlingame, CA) for nuclear visualization. The incorporation of DiI-Ac-LDL was visualized using a Nikon Eclipse E600 fluorescent microscope (Nikon, Kanagawa, Japan) equipped with Leica DC 300F (Leica Microsystems DI, Cambridge, UK) and Color View III (Olympus, http://www.olympus-global.com/en/) digital cameras. Pictures were taken by Adobe Photoshop 7.0 (Adobe Systems Norge AS, Oslo, Norway) and Cell-B software (Olympus).
In Vitro Tube Forming Assay
Matrigel Basement Membrane Matrix (BD Biosciences) was used as a substrate for vasculogenesis assays to determine the functional ability of the isolated cells to form networks. Matrigel (50 μl) was spread at the bottom of 96-well plates (Costar, Corning, New York, NY) and used as a coat upon which freshly isolated (uncultured) and cultured (P2, P6, and P12) AT-ECs were seeded in 100 μl medium. Human dermal MVECs (P6) were used as a positive control. AT-MSCs alone and mixed with AT-ECs were also tested in this assay. Cultures were incubated at 37°C in a humidified incubator with 5% CO2 for 16–20 h. The presence of tubes was visualized using a Leica DMIL inverse microscope (Leica Microsystems, Wetzlar, Germany). The assay was performed in triplicates. PKH67 Fluorescent Cell Linker Kit was used to label the cells for the fluorescence image. Images were taken with a Leica DFC 320 camera (Leica Microsystems) using Adobe Photoshop 7.0 software.
In Vivo Vasculogenesis Assay
AT-ECs from donor 2 at 0.3 times; 106, 0.5 times; 106, and 2 times; 106, alone or combined with AT-MSCs from the same donor (4:1 ratio), were resuspended in 50 μl PBS, mixed with 200 μl of Matrigel on ice, and injected subcutaneously into 8-week-old nonobese diabetes/severe combined immunodeficiency (NOD/SCID) mice. Each experimental condition was performed with two mice. Animal experiments were approved by the National Animal Research Authority.
The Matrigel plugs were removed after 6 weeks, fixed in 10% PFA (Electron Microscopy Sciences, Hatfield, PA) overnight, embedded in paraffin, and sectioned. For evaluation of the angiogenic potential, 3-μm-thick sections were stained for morphological evaluation using hematoxylin and eosin (data not shown). For immunohistochemistry analysis sections were deparaffinized and then boiled in PBS containing 0.05% citraconic anhydride (pH 7.4) for 20 min for epitope retrieval. Mouse anti-human CD31 primary antibody at 1:20 (DAKO) was applied overnight in a humidified chamber at 4°C diluted in PBS containing 1.25% bovine serum albumin (BSA) and 0.1% saponin (SERVA Electrophoresis GmbH, Heidelberg, Germany) for blocking and permeabilization. Slides were washed three times in PBS. Human vessels were detected using cyanine 3 (CY3)-conjugated donkey anti-mouse IgG (1:600; Jackson ImmunoResearch) at room temperature for 1.5 h. Unbound antibody was removed with three washes in PBS. Double staining with fluorescein isothiocyanate (FITC) conjugated UEA-1 at 1:200 was performed together with the secondary antibody incubation step. As a control, secondary antibody alone was used to assess nonspecific fluorescence.
Data Analysis
Data are shown as mean ± SEM and determined using GraphPad Prism version 5 software.
Results
Morphology and In Vitro Growth Characteristics of AT-ECs
Because AT-ECs are abundant within the SVF, even after purging of non-endothelial cells, we could obtain 15–60 × 106 cells from 300–600 ml liposuction material, of which 8–10 × 106 were seeded for further analysis. Cell yield was 67.2 × 103 ± 20.3 × 103 (mean ± SEM) cells/ml of lipoaspirate.
AT-EC colonies emerged after 4–5 days in the culture. The cultures exhibited the classical EC cobblestone morphology from P1 and maintained their morphological characteristics for as long as cell proliferation continued (Fig. 1A). AT-ECs quickly entered a phase of logarithmic growth (Fig. 1B). During this phase, the population doubling time was 2.3 days. The proportion of AT-ECs that were able to attach and form colonies was difficult to quantify. However, when the cells were passaged for the first time, after 8 days, the cell number was unchanged (two donors) and doubled (one donor). Considering the population doubling time, this suggests that approximately half of the cells attached and formed colonies. Logarithmic growth ceased for cells from two donors at P14, and for one donor at P19.
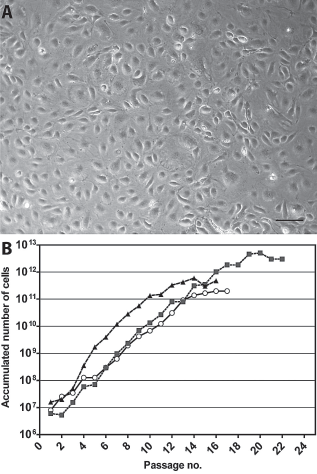
Growth kinetics and phase contrast morphology of adipose tissue-derived endothelial cells (AT-ECs). (A) Light microscopy of a confluent monolayer of AT-ECs (passage 8) exhibiting typical cobblestone morphology. Cultures were examined by a Leica DMIL inverted microscope. Image was taken with a Leica DFC 320 camera using Adobe Photoshop 7.0 software and a 10×/0.22 Leica objective lens. Scale bar: 100 μm. (B) Calculated accumulated cell counts from cultures of AT-ECs isolated from three different donors.
Gene Expression in AT-ECs
The endothelial nature of these cells was examined in a number of different ways. Figure 2 shows the expression of genes relevant to the endothelial lineage and to the mesenchymal lineage, as AT-MSCs were also present in SVF and could be contaminating the AT-ECs. The expression of these genes was calculated relative to the expression of the endogenous control gene CDKN1A. Relative to the expression in uncultured AT-ECs, the mRNA levels for platelet endothelial cell adhesion molecule-1 (PECAM1), vascular endothelial cadherin (CDH5), vascular endothelial growth factor receptor 2 (KDR) and von Willebrand factor (VWF) were uniformly upregulated in cultured cells at P2, and then remained stably expressed. CD34, CD90 (THY1) and endothelial nitric oxide synthase (NOS3) showed some between-donor variation, but the trend was for an overall relatively stable expression of these genes. The mRNA expression levels of the endothelial lineage genes in the cultured AT-ECs were similar to the commercially available human MVECs whereas the expression levels of THY1 and CD44 were higher in the AT-ECs (Fig. 3A). In human chondrocytes, which were used as a negative control, endothelial markers PECAM1, CD34, NOS3, CDH5, KDR, and VWF were not expressed. However, CD44 and THY1 were expressed at high levels (data not shown).

Real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis. Graphs represents expression of mRNA encoding endothelial markers CD31/PECAM1, CD34, VEGFR-2/KDR, VWF, NOS3, VE-cadherin/CDH5, and mesenchymal stem cell (MSC) markers CD90/THY1 and CD44 at different passages (P0 = uncultured cells; P2 = passage 2; P6 = passage 6; P12 = passage 12) in three donors. Graphs were created using GraphPad Prism 5 software.

Gene expression, in vitro vasculogenic potential, and cell surface marker expression of primary adult human dermal microvascular endothelial cells (MVECs). (A) Real-time RT-PCR analysis. Graphs represents expression of mRNA encoding endothelial markers CD31/PECAM1, CD34, VEGFR-2/KDR, VWF, NOS3, VE-cadherin/CDH5, and MSC markers CD90/THY1 and CD44 at passage 6. Graphs were created using GraphPad Prism 5 software. (B) In vitro vasculogenesis assay. MVECs (P6) create extensive capillary-like structures in Matrigel. Vascular network was examined after 20 h by a Leica DMIL inverted microscope. Image was taken with a Leica DFC 320 camera using Adobe Photoshop 7.0 software and a 10×/0.22 Leica objective lens. Scale bar: 100 μm. (C) Flow cytometric analysis. Flow cytometry histograms of cells from passage 6. Gray histograms represent cells stained with fluorescent antibodies conjugated to fluorescein isothiocyanate (FITC), allophycocyanin (APC), phycoerythrin (PE), and phycoerythrin-cyanin 5 (PE-Cy5). Isotype-matched control antibodies are overlaid in a black line on each histogram.
Flow Cytometry Analysis
To examine the efficiency of our isolation technique and possible phenotypic changes due to in vitro expansion, flow cytometry analysis was performed. Figure 4A shows the distribution of the CD31+, CD44+, CD45+, and CD90+ cell populations in the SVF. Figure 4B shows that after the removal of CD44+ cells from the SVF, 4% of CD44+ cells remained in the cell suspension. Of the CD44– cells, 80% were CD31+ and 87% were CD90+. However, despite the low proportion of CD44+ cells present after the initial isolation, some of the cultured cells appeared as fibroblastoid toward the first passage. Therefore, to minimize the contamination by non-endothelial cells, we repeated the removal of CD44+ cells at the first passage. This resulted in a >95% CD31+ endothelial cell culture with <2% CD44+ cells contaminating the AT-EC culture at P2. In the course of cell culture, an increasing proportion of the AT-ECs expressed CD44, so that for this donor, 77% of the cells were CD44+ at P12, despite retaining cobblestone morphology and being almost uniformly CD31+. This observation was made for cells from all the donors, and substantiated the increasing expression of CD44 mRNA shown in Figure 2.

Flow cytometric analysis. (A) Cell surface marker expression of the stromal vascular fraction (SVF). (B) Purity and phenotype change of AT-ECs at different passages. Flow cytometry plots of cells from donor 3 (A) and donor 2 (B), representative of the three donors. Cells were costained for CD31 (platelet endothelial cell adhesion molecule, PECAM), CD90 (Thy-1), CD44 (homing-associated cell adhesion molecule, H-CAM), and CD45. Isotype-matched control fluorescent antibodies were used as negative controls.
Of the uncultured CD31+ cells, 90% were also strongly CD90+. However, the surface expression of CD90 was quickly lost, so that at P2 hardly any surface expression of CD90 was observed for any of the donors (Fig. 4B). Interestingly this was not a reflection of the mRNA expression for THY1, which was maintained practically unchanged through P6, and also through P12 for two of the donors (Fig. 2). A similar observation was made for CD34, which was expressed on the surface of most of the uncultured cells, but then quickly and persistently disappeared from the surface (Fig. 5A). At the mRNA level, expression was maintained for two of the donors, and reduced at P6 and P12 for the last donor.

Flow cytometric characterization of AT-ECs. Flow cytometry histograms of cells from donor 3, representative of the three donors at different passages. Gray histograms represent cells stained with fluorescent antibodies conjugated to (A) fluorescein isothiocyanate (FITC), (B) allophycocyanin (APC), (C) phycoerythrin (PE), and (D) phycoerythrin-cyanin 5 (PE-Cy5). Isotype-matched control antibodies are overlaid in a black line on each histogram.
Figure 5 shows that uncultured AT-EC express endothelial markers such as CD34, CD144, CD146, CD105, human lymphocyte antigen (HLA) DR, CD54, CD102, vWF, and UEA-1. The expression of most of these molecules was high and persistent in the cultured cells. However, for HLA DR the expression disappeared at P2, and for CD144 there was a tendency to reduced expression in cultured cells. Interestingly this observation, too, was in contrast to the changes observed for CD144 (CDH5) at the mRNA level, where expression was higher in the cultured cells (Fig. 2). AT-ECs were uniformly negative for CD133/1 and CD133/2, which have been used as markers for EPCs (3). They expressed moderate levels of VEGFR-1 and VEGFR-2 at the uncultured state. The expression of VEGFR-1 was reduced or absent in the cultured cells, while VEGFR-2 was upregulated both at the protein and the mRNA level in these cells. The cells were negative at all times for the hematopoietic surface antigens CD45, CD14, CD19, and CD3 (data not shown).
Human dermal MVECs were used as a positive control for the examination of the endothelial and mesenchymal cell surface antigen expression (Fig. 3C). As expected there were some differences in the surface marker expression between the two cell populations. The MVECs did not express CD44 during the culture, whereas CD34 was expressed on the surface of a proportion of the cells.
DiI-Ac-LDL Uptake
Figure 6 shows that AT-ECs take up DiI-Ac-LDL (Fig. 6A). This characteristic is common for ECs and further indicates the endothelial properties of the cultured cells. Human chondrocytes and AT-MSCs did not take up DiI-Ac-LDL (Fig. 6B, C, respectively).
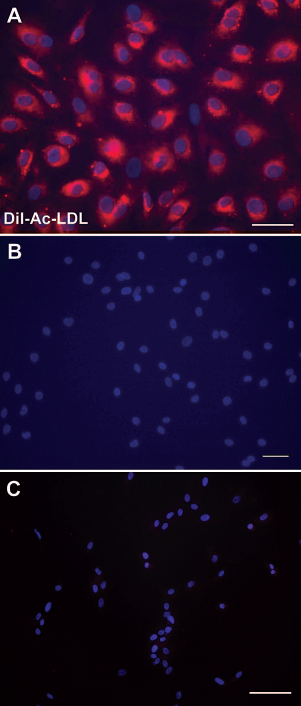
DiI complex acetylated low-density lipoprotein (DiI-Ac-LDL) uptake. (A) Uptake of DiI-Ac-LDL by AT-ECs and (B) by human chondrocytes and (C) adipose tissue-derived mesenchymal stem cells (AT-MSCs) used as negative controls. Scale bars: 50 μm (A) and 100 μm (B, C). 4′,6-Diamidino-2-phenylindole (DAPI) stains the nuclei in blue in each image. Slides were assessed using a Nikon, Eclipse E600 fluorescent microscope equipped with Nikon 20x/0.45 and 40x/0.75 objective lenses and Leica DC 300F and Color View III digital cameras. Images were taken using Adobe Photoshop 7.0 and Cell B software.
In Vitro and In Vivo Vasculogenesis Assays
The freshly isolated cells did not form vascular tubes when seeded on Matrigel (Fig. 7A), while the cultured cells both at early (Fig. 7B, C) and late passage (Fig. 7D) formed capillary-like structures, similar to the commercially obtained MVECs (Fig. 3B). AT-MSCs alone (Fig. 7E) or AT-MSCs mixed with AT-ECs (Fig. 7F) formed single tube-like structures, but not the extensive vascular network formed by the AT-ECs alone.

In vitro vasculogenesis assay. Representative images of vascular network formation on Matrigel. (A) Freshly isolated AT-ECs are not able to form vascular tubes while seeded on Matrigel. (B) Cultured AT-ECs (P2) create extensive capillary-like structures in Matrigel. (C) Image showing tube formation ability of AT-ECs (P2) following PKH67 fluorescent cell labeling of AT-ECs. (D) Extensive capillary-like network formed by late passage AT-ECs (P13). (E) AT-MSCs and (F) AT-MSCs mixed with AT-ECs were not able to form capillary-like networks in Matrigel. Vascular network was examined after 20 h by a Leica DMIL inverted microscope. Images were taken with a Leica DFC 320 camera using Adobe Photoshop 7.0 software and 4×/0.10 and 10×/0.22 Leica objective lenses. Scale bars: 50 μm (B) and 100 μm (A, C, D, E, F).
To determine if AT-ECs could form functional blood vessels in vivo, 0.3 × 106, 0.5 × 106, and 2 × 106 of AT-ECs were mixed with Matrigel and implanted into NOD/SCID mice. In addition, because MSCs have been shown to be identical to pericytes in vivo and thus might improve the vasculogenic properties of the AT-ECs, AT-MSCs from the same donor were mixed into the Matrigel in half of these experiments. The implants were removed and evaluated at 6 weeks. Where AT-ECs were applied at 0.5 × 106 and 2 × 106 per Matrigel, either alone or combined with AT-MSCs, tubular vessel-like structures could be identified (Fig. 8). The degree of vascular network formation tended to be greater when combined cell populations were applied. The vessels were human-derived, as they stained with antibodies specifically recognizing human CD31 and UEA-1 (Fig. 8A–F). The presence of mouse red blood cells in the lumen indicates that functional capillary networks were formed between the host vasculature and the human-specific luminal structures created by the implanted cells. Injection of the Matrigel alone resulted in no vascular network formation (data not shown). The specificity of the human CD31 antibody and UEA-1 lectin was confirmed by negative staining of mouse aorta (data not shown).

In vivo vasculogenic potential of AT-ECs. (A, D) Representative fluorescence images of human specific CD31 (Cy3) and (B, E) human-specific Ulex europaeus agglutinin I lectin (UEA-1) (FITC) staining at 400× and 1000× magnification. (C, F) Merged images of CD31 and UEA-1 staining. Scale bars: 50 and 10 μm, respectively. DAPI stains the nuclei in blue in each image. White arrows indicate the position of the vascular lumen and the endothelium. Mouse red blood cells within the vascular lumen are seen to emit both red and green autofluorescence. Slides were assessed using a Nikon Eclipse E600 fluorescent microscope equipped with Nikon 40×/0.75 and 100×/1.30 oil objective lenses and a Leica DC 300F digital camera. Images were taken using Adobe Photoshop 7.0 software. All images were assembled into multipanel figures using Adobe Illustrator CS4 (Adobe Systems Norge AS, Oslo, Norway).
Discussion
Tissue neovascularization may in itself treat diseases such as myocardial and peripherovascular ischemia, and it is increasingly recognized as an essential ingredient for the successful engineering and transplantation of bone, liver, and a range of other tissues (11,27,28,32,36,44,47). Neovascularization will not occur in the absence of ECs. Classical sources of human ECs have been umbilical vein (HUVEC) and MVECs from foreskin (8,15,16). However, in a therapeutic setting these cells will almost always be allogeneic to the patient. A new paradigm was established with the identification of EPCs (2). Human EPCs can be isolated from the blood and expanded to clinically relevant numbers, but this requires extended in vitro culture (24,34). Recently, MSCs have been identified as important for neovascularization because they seem to have a role as pericytes in most tissues (7). Thus, the establishment of robust tissue vascularization may require at least two cell types: ECs and MSCs/pericytes. We have previously purified and characterized MSCs from AT, and have shown how they can be obtained as primary uncultured cells in large numbers (4). The present study identifies AT as a source also of ECs. AT-ECs, too, can be obtained in large numbers as primary uncultured cells, and they, too, are readily expanded in vitro. The negative selection strategy allows the isolation of AT-ECs with high purity, and with no contamination in the cell culture of the immunobeads used for selection. As AT is easily obtainable from all patients, an autologous source of the most important cells required for vasculogenesis should now be readily available in most therapeutic situations.
It is a little difficult to place the AT-ECs within the established EC hierarchy. As they do not express either isoform of CD133, and do express CD144 (VE-cadherin) and vWF, they are not early EPCs (13,43). At the other end of the hierarchy, human MVECs have been shown to strongly express CD141 (3), which was only weakly or not expressed on the cells characterized here. AT-ECs most resemble late EPCs based on the phenotype CD34+CD133–vWF+CD144+VEGFR-2+eNOS+CD31+ (13,43). However, if almost unlimited in vitro expansion potential is required for cells to deserve the “progenitor” designation, then the AT-ECs are not EPCs. This was shown by Melero-Martin et al. to be a property of human blood-derived endothelial progenitor cells (24). In contrast, our AT-ECs ceased to proliferate after 14–19 passages. Consequently, these cells are most likely differentiated ECs with high in vitro proliferative capacity.
Although we were able to isolate high numbers of primary AT-ECs, in vitro expansion was necessary to obtain the number of cells that will likely be needed in most protocols of in vivo vasculogenesis (21,25,29,35,48). For instance, 108 cells could be obtained after only a few passages. At this time the cells were in a log phase of growth and showed the classical cobblestone morphology, but certain changes had already been induced by in vitro culture. On the cell surface, CD34, CD90, and HLA-DR had quickly disappeared. Interestingly, CD34 and THY1 (CD90) mRNA were maintained for some time after the cell surface expression was lost, suggesting that these phenotypes might possibly be induced under other culture conditions. For comparison, CD90 and HLA-DR were also found not to be expressed on blood-derived human EPCs, while CD34 was reported to be expressed on these cultured cells (24,34). Equally strikingly, CD44 changed from being practically absent on the uncultured AT-ECs, which were depleted for CD44-expressing cells, to being gradually more expressed. The depletion procedure was sufficiently efficient to strongly suggest that this was a bona fide change in phenotype, and not an outgrowth of a small subset with an otherwise similar phenotype. At P12, a very large proportion of the cells expressed CD44. A similar, although less dramatic, change was noted for VEGFR-2 and vWF. For both these markers a parallel increase was noted at the mRNA level.
The functional properties of the AT-ECs were examined using vasculogenesis assays. In vitro, the AT-ECs formed tubes similar to the commercially obtained MVECs used for positive control. Interestingly, we also observed very efficient tube formation using cells obtained from late passages, suggesting that the AT-ECs retain their functional capacity better than cord blood-derived EPCs, which were found to lose functional capacity upon in vitro passaging (24). To our surprise, uncultured AT-ECs did not form tubes in Matrigel in vitro. To the best of our knowledge, this assay has not been performed on any population of uncultured ECs before, presumably because the different primary ECs populations described to date have been too few to perform assays of this nature. While it is likely that the AT-ECs, given the right stimulus, would be able to create new vessels in vivo, the in vitro vasculogenesis assay presented here may be able to elicit vasculogenic properties only from the in vitro cultured ECs to which it has been adapted. Results from the in vivo blood vessel forming assay showed that AT-ECs alone were able to form vessels containing mouse-derived erythrocytes. However, these results were inconsistent, and in all cases the vessel formation was denser and more consistent when AT-MSCs were added to the Matrigel. More experiments must be performed, perhaps also using in vivo ischemia models of neovascularization, in order to support these observations with statistics. It makes sense, though, to think that addition of pericytes to ECs will yield more robust blood vessels. After all, these cultured cells probably derive from the very cells that made up the layers of the walls of the small vessels in the AT from which they were isolated (6,42). Hopefully this combination of autologous cells may, in the future, be shown to provide blood supply to ischemic tissues in vivo or a vascular network for in vitro engineered tissue implants.
Footnotes
Acknowledgments
The authors thank Colosseum Klinikken, Oslo (Tone Irene Langslet) for kindly providing liposuction material, Linda T. Dorg and Finn P. Reinholt for sectioning and H&E staining, Peter Hofgaard and Bjarne Bogen for help with the animal experiments, Axel M. Küchler for helpful suggestions pertaining to immunohistochemistry, and Steven Hoel for advice, support and discussions. This work was supported by a Storforsk grant from the Norwegian Research Council. The authors declare no competing financial interest.