
Abstract
Adipose tissue may represent a potential source of adult stem cells for tissue engineering applications in veterinary medicine. It can be obtained in large quantities, under local anesthesia, and with minimal discomfort. In this study, canine adipose tissue was obtained by biopsy from subcutaneous adipose tissue or by suction-assisted lipectomy (i.e., liposuction). Adipose tissue was processed to obtain a fibroblast-like population of cells similar to human adipose-derived stem cells (hASCs). These canine adipose-derived stem cells (cASCs) can be maintained in vitro for extended periods with stable population doubling and low levels of senescence. Immunofluorescence and flow cytometry show that the majority of cASCs are of mesodermal or mesenchymal origin. cASCs are able to differentiate in vitro into adipogenic, chondrogenic, myogenic, and osteogenic cells in the presence of lineage-specific induction factors. In conclusion, like human lipoaspirate, canine adipose tissue may also contain multipotent cells and represent an important stem cell source both for veterinary cell therapy as well as preclinical studies.
Introduction
A promising application in the emerging field of veterinary regenerative medicine and surgery is cell therapy, rendering the isolation and characterization of stem cells from a variety of sources areas of great interest.
An abundant and accessible source of stem cells is adipose tissue. These cells, called adipose-derived stromal cells (ASCs), are fibroblast-like cells capable of multipotential differentiation, which have been found in different species (4,27,29, 35). Several groups have demonstrated that human mesenchymal cells within the stromal-vascular fraction (SVF) of subcutaneous adipose tissue [processed lipoaspirate (PLA) cells] are capable of differentiation in multiple lineages, including myocytes, in the presence of lineage-specific inductive media (2,5,8–10,15,16,19,20,22,23,25,34,35).
In humans, ASCs for autologous transplantation are isolated relatively quickly from adipose tissue by collagenase digestion (6). We have recently shown that ASCs from human subcutaneous fat were able to differentiate in adipogenic, osteogenic, chondrogenic, and myogenic lineages and produce human muscle proteins in vitro and in vivo (30,31).
Successful transplantation of canine adipose-derived stem cells (cACSs) in dogs was reported by Li et al. (12) and Black et al. (1). However, these manuscripts lacked the full characterization of the administered cell population. Here we report the isolation, characterization, and multilineage differentiation potential of cASCs from subcutaneous adipose tissue by liposuction and biopsy procedures.
Materials and Methods
All experimental protocols were approved by the ethics committee on animal use from the Institute of Biosciences, University of São Paulo. For this study, adipose tissue was collected from normal golden retriever dogs from the Brazilian Colony of Golden Retriever Muscular Dystrophy, Faculty of Veterinary Medicine and Zoo-tecny, University of São Paulo. Subcutaneous adipose tissue was collected from the area over the dorsal gluteal muscles of 10 dogs (aged 4 months to 4 years).
Adipose Tissue Harvesting
Dogs were sedated upon intramuscular (IM) injection with meperidine (2 mg/kg) and acetylpromazine (0.05 mg/kg). The area over the dorsal gluteal muscles was asceptically prepared, and skin and subcutaneous tissues were desensitized by local infiltration of 2% lidocaine (Fig. 1A). A 0.5–1.0-cm incision was made parallel to the vertebral column. The liposuction procedure was performed by injecting infiltrate containing the vasoconstrictor epinephrine. Then adipose tissue was removed from the subcutaneous space by means of blunt-tip hollow cannula attached to a syringe at negative pressure (Fig. 1B). About 15 ml of adipose tissue was harvested over the superficial gluteal fascia for immediate cASC isolation and the skin incision apposed with nylon sutures (Fig. 1C). Adipose tissue biopsies were performed under local anesthesia. A 1–2-cm incision was made and the subcutaneous adipose tissue was collected (Fig. 1D) and the incision was closed with nylon sutures (Fig. 1E).

Adipose tissue harvest. (A) The area over the dorsal gluteal muscles prepared for the procedure. (B) Liposuction procedure. (C) Skin incision after the liposuction. (D) Subcutaneous adipose tissue biopsy. (E) Skin incision after the biopsy.
cASC Isolation and Expansion
Cells were isolated using modified methods previously described (7). Briefly, the adipose tissue was washed extensively with equal volumes of PBS containing antibiotics (100 U/ml of penicillin and 100 g/ml of streptomycin). The infranatant containing hemopoietic cells suspended in PBS was removed. Then the tissue was dissociated with 0.075% collagenase (Sigma) for 15 min. Enzyme activity was neutralized with Dulbecco's modified Eagle's media- high glucose (DMEM-HG; Gibco) containing 10% FBS (Gibco). The infranatant was centrifuged at 1200 × g for 5 min to pellet the cells. The cells from the pellet SVF were filtered to remove debris and seeded in tissue culture plates (NUNC) at 1,000-3,500 cells/cm2 in DMEM-HG 10% FBS. Cultures were washed with PBS 24–48 h after plating to remove unattached cells and fed with fresh media.
The cultures were maintained at 37°C with 5% CO2 in growth media (GM-DMEM-HG 10% FBS). When they achieved about 70% confluence, the cells were trypsinised (0.025%, Invitrogen) and plated at a density of 5,000/cm2. Cultures were passaged repeatedly after achieving a density of 70–80%. The remaining cells were cryopreserved in cryopreservation media (10% dimethylsulfoxide, 10% DMEM-HG, 80% FBS), frozen at −80°C in an isopropanol-jacketed closed container, and stored in liquid nitrogen the next day.
Multilineage Differentiation
Cells were analyzed for their capacity to differentiate into adipogenic, chondrogenic, osteogenic, and myogenic lineages as described in Zuk et al. (35).
Adipogenic Differentiation
Subconfluent cells were cultured in GM supplemented with 1 μM dexamethasone (Sigma), 500 μM 3-isobutyl-1-methyl-xanthine (IBMX, Sigma), 60 μM indomethacin (Sigma), and 5 μg/ml insulin (Sigma). Adipogenic differentiation was confirmed on day 21 by intracellular accumulation of lipid-rich vacuoles stainable with Oil Red O (Sigma). For the Oil Red O stain, cells were fixed with 4% paraformaldehyde for 30 min, washed, and stained with a working solution of 0.16% Oil Red O for 20 min.
Chondrogenic Differentiation
Subconfluent cells were cultured in chondrogenic differentiation medium consisting of DMEM-low glucose supplemented with 100 nM dexamethasone, 50 μM ascorbic acid-2 phosphate (Sigma), 1 mM sodium pyruvate (Gibco), 10 ng/ml TGF-β1 (R&D Systems), and 1% ITS-Premix (Becton Dickinson). Medium was changed every 3–4 days, and cells were fixed on day 21 with 4% paraformaldehyde (PFA). Chondrogenesis was demonstrated by staining with toluidine blue and immunofluorescence using anti-collagen type II antibody (1:100, Abcam).
Osteogenic Differentiation
To promote osteogenic differentiation, subconfluent cells were treated with GM supplemented with 50 μM ascorbate-2 phosphate, 10 mM β-glycerophosphate (Sigma), and 0.1 μM dexamethasone for 21 days. Osteogenesis was demonstrated by accumulation of mineralized calcium phosphate assessed by von Kossa stain. Briefly, cells were stained with 1% silver nitrate (Sigma) for 45 min under ultraviolet light, followed by 3% sodium thiosulphate (Sigma) for 5 min, and then counterstained with van Gieson.
Myogenic Differentiation
For myogenic differentiation, cASCs cells were cultured in GM supplemented with 0.1 μM dexamethasone (Sigma), 50 μM hydrocortisone (Sigma), and 5% horse serum (Gibco) for 45 days. After that cells were labeled with anti-myosin (1: 100, Sigma).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde in PBS for 20 min at 4°C, permeabilized in 0.05% Triton X-100 in PBS for 5 min. Nonspecific binding was blocked with 10% FBS in PBS for 1 h at room temperature. Cells were incubated with primary antibody (1:100) overnight at 4°C. After several washes, cells were incubated with secondary (1:100, Sigma) antibodies against mouse IgG tagged with Cyanine 3 (Cy3; red) for 2 h at room temperature. Slides were counterstained with DAPI (4′-6-diamidino-2-phenylindole, Sigma). All images in the same set (samples and controls) were obtained using the same photographic parameters of exposition and speed. Images were captured using the Axiovision 3.0 image analysis system (Carl Zeiss).
Flow Cytometry
Cells were evaluated for cell surface protein expression using flow cytometry. The flow cytometry was performed on Guava EasyCyte System (Guava Technologies) using a blue laser (488 nm). Cells were pelleted, resuspended in PBS at a concentration of 1 × 105 cells/μl, and stained with saturating concentration of antibodies. Cells were incubated in the dark for 45 min at room temperature. After incubation, cells were washed three times with PBS and resuspended in 0.25 ml of cold PBS. Cell viability was accessed with Guava ViaCount reagent (Guava Technologies).
cASCs were incubated with the following primary antibodies: CD13-PE, CD29-PECy5, CD31-PE, CD34-PE, CD44-FITC, CD45, CD73, CD90-PE, CD105 e CD117-PECy5 (Becton Dickinson). The following antibodies have been raised against human cells: CD13, CD71, and CD105. Unconjugated markers were treated with anti-mouse PE secondary antibody (Guava Technologies).
Flow cytometer settings were established using unstained cells. Cells were gated by forward scatter to eliminate debris. To eliminate the possible autofluorescence of cASCs, we removed the contribution of unstained cells in the measurement channel. A minimum of 10,000 events was counted for each analysis.
RNA Isolation and Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Total RNA was harvested from cultured cells using Tryzol (Invitrogen) following manufacturer's instructions. The RNA was treated with DNase (Invitrogen). A total of 1 μg of total RNA was reverse-transcribed with SuperScript™ III First-Strand Synthesis System (Invitrogen). All amplifications were performed in an MJ Research PTC-200 thermocycler (MJ Research) for 24 cycles after the initial 2-min denaturation at 94°C. The PCR primers are listed in Table 1. The PCR products were separated on 6% polyacrylamide gel by electrophoresis, stained with ethidium bromide, and visualized under UV light. Digital images were captured with ImageQuant (GE Healthcare).
PCR Primers

cASCs Transduction with Lentivirus Vector
The visualization of cASCs cells for in vitro and in vivo studies can be improved if done with GFP-positive cells. For this purpose we transducted cASCs with GFP lentivirus.
Supernatant containing the FUGW lentivirus (13) was produced as described previously by Strauss et al. (26) and concentrated by ultracentrifugation. Undifferentiated cASCs at passage 2 were incubated at 37°C, in a six-well plate (Nunc), using a minimal volume of GM in the presence of vector particles (20 PFU/cell) and 8 μg/ml Polybrene (Sigma). After 4 h, 2 ml of GM was added and the media was changed the next day.
Karyotype Analysis
For evaluation of any chromosomal abnormality at latter passages, chromosome preparations were performed in cASC cultures. Briefly, metaphase cells were arrested with 0.1 μg/ml colchicine (Sigma) for 20 min. Then, cASCs were detached from cultures flasks using TrypLE (Gibco), resuspended in a hypotonic solution (0.075M KCl), and incubated for 20 min at 37°C. Cells were pelleted at 1000 rpm for 10 min and fixed by washing three times in methanol/glacial acetic acid (3:1). Chromosome spreads were obtained by pipetting suspension drops onto clean glass slides and air dried. The best metaphases were captured with Axioplan 2 microscope (Zeiss) and analyzed with Ikaros 3 software (Zeiss).
Results
Characterization of cASCs
cASC cultures were maintained in DMEM supplemented with 10% FBS. Supplementation with FBS has been shown to be important for human ASC attachment and proliferation in vitro (35). We observed that cASCs are easy to expand in vitro and show a fibroblast-like morphology, consistent with that of human ASCs (Fig. 2A–D). At both early and late passages, cells maintained a diploid karyotype of 78 chromosomes (Fig. 2E).

Typical morphology of cASCs. (A) Forty-five minutes after the establishment of the culture. Some cells remain in the supernatant. Scale bar: 200 μm. (B) cASCs at passage 3. Scale bar: 200 μm. (C) cASCs at passage 4: cASCs morphology is similar to that found in human ASCs. Scale bar: 100 μm. (D) High-density ASCs culture at passage 4. Scale bar: 200 μm. (E) Karyotype of cASC cell lineage after 10 passages, showing an euploid number of chromosomes.
cASCs from four unrelated dogs were characterized by flow cytometry for the expression of 10 cell surface proteins (CD13, CD29, CD31, CD34, CD44, CD45, CD73, CD90, CD105, and CD117). Cell viability was above 96% by Guava ViaCount reagent (Guava Technologies).
At passage 4, the majority of cASCs expressed CD44, CD29 (β1 integrin) and CD90 (Thy1) adhesion molecules. Other markers, including CD14, CD34, CD45, and CD117, were consistently absent or expressed in few cells (Fig. 3). Interestingly, CD13, CD105, and CD73, known to be positively expressed in human ASCs, were negative in the canine ASC population, which might be explained by the nonspecific staining of human antibodies in canine cells. As surface markers are not sufficient for the identification or definition of mesenchymal stem cell (MSC), cASCs were subjected to differentiation studies for further confirmation of their MSC property.
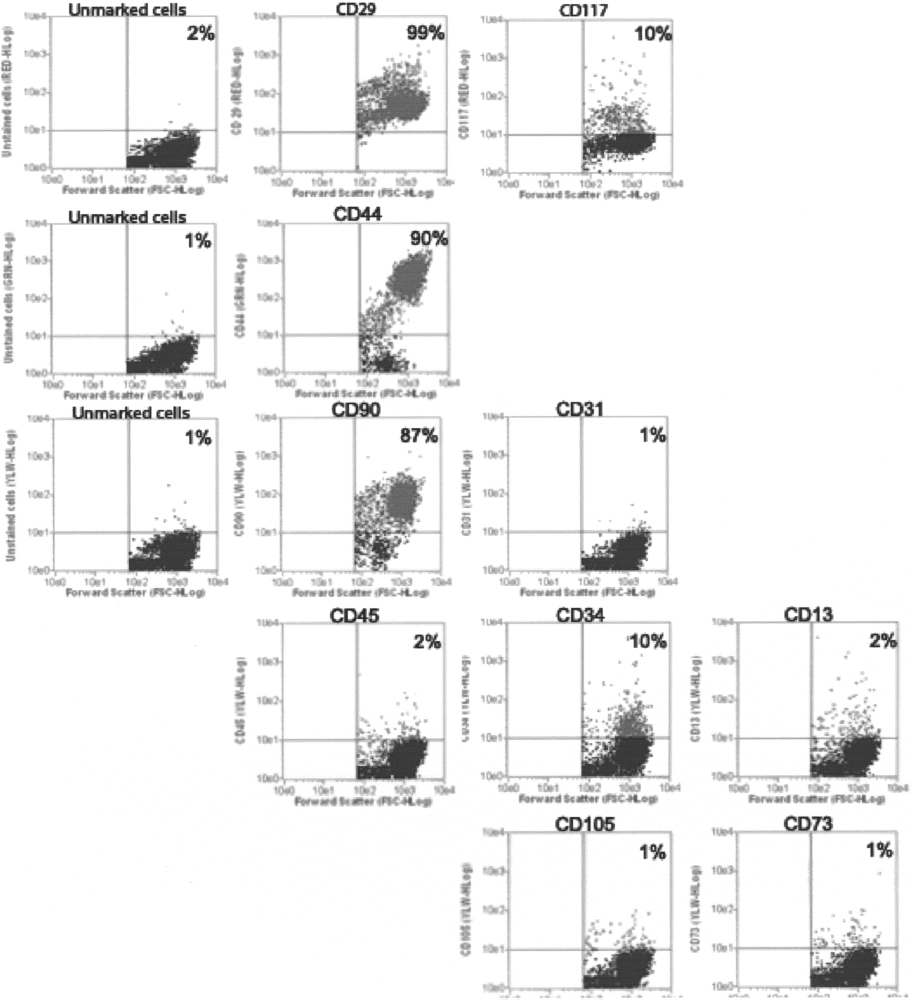
Immunophenotyping of ASCs at passage 4. Values represent the mean percentage of positively stained cells as analyzed by flow cytometry. Graphs show forward scatter versus fluorescence intensity of the indicated antigen.
The plasticity of cASCs was assessed after lineage induction. Myogenic, adipogenic, chondrogenic, and osteogenic differentiation was demonstrated by the expression of myogenic markers (myosin), lipid vacuoles, mucopolysaccharide-rich extracellular matrix, and calcium deposits, respectively (Fig. 4) and by the expression of tissue-specific mRNAs (Fig. 5). These results confirmed the mesenchymal nature of the isolated cells and their multipotent potential.

Differentiation potential of cASCs at passage 4. (A) The adipogenic differentiation was detected by the formation of intracytoplasmic lipid droplets stained with Oil Red O. Scale bar: 200 μm. (B) Undifferentiated ASCs stained with Oil Red O. Scale bar: 200 μm. (C) cASCs after induction with adipogenic media still show GFP expression. Scale bar: 200 μm. (D) Osteogenic differentiation was demonstrated by calcium deposition shown by von Kossa stain. Scale bar: 200 μm. (E) Undifferentiated ASCs stained with Von Kossa. Scale bar: 200 μm. (F) cASCs after induction with osteogenic media still show GFP expression. Scale bar: 200 μm. (G) Chondrogenic differentiation in monolayer culture was demonstrated by staining with toluidine blue. Scale bar: 200 μm. (H) Undifferentiated ASCs stained with toluidine blue. Scale bar: 200 μm. (I) cASCs after induction with chondrogenic media still show GFP expression. Scale bar: 200 μm. (J) Chondrogenic differentiated cells labeled with anti-collagen type II antibody. Scale bar, 200 μm. (K) Undifferentiated ASCs labeled with anti-collagen type II antibody. Scale bar: 200 μm. (L) Myogenic differentiation was assessed by immunofluorescence. Induced cells were labeled with anti-myosin monoclonal antibody. Scale bar: 50 μm. (M) Undifferentiated ASCs labeled with antihuman myosin monoclonal antibody. Scale bar: 50 μm. (N) cASCs after induction with myogenic media still show GFP expression. Scale bar: 50 μm.

mRNA expression of specific differentiation markers. (A) Myogenic markers: myogenin (Myog), dystrophin (Dyst) and MyoD. (B) Adipogenic markers; FABP4, leptil, and LPL. (C) Osteogenic markers: osteopontin (OPN), COL1A1, bone sialoprotein (BSP). (D) Chondrogenic markers; COL2A, SOX9, and aggrecan (AGC). The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as reference for evaluating the quality of mRNA.
Adipogenesis
cASCs showed a rounder shape after 7 days in adipogenic medium. Two weeks after initial induction, the adipogenic differentiation was confirmed by Oil Red O staining of lipid droplets present throughout the cytoplasm (Fig. 4A). Expression of FABP4 and LPL was seen only in adipo-induced cells (Fig. 5B). On the other hand, basal level of leptin mRNA was observed in noninduced control cells, and the expression level was increased following adipogenic induction.
Osteogenesis
cASCs exposed to osteogenic medium exhibited changes in cell morphology after 5 days in culture, showing a polygonal form. Mineralized nodular structures appeared in 1 or 2 weeks and were assessed by von Kossa Stain, which localized the calcium deposits (Fig. 4C). Expression of osteopontin, COL1A1, and BSP was observed only in induced cells, with no basal expression in control cells (Fig. 5C).
Chondrogenic Differentiation
After 21 days cultured in chondrogenic medium, cASCs cells were stained with toluidine blue, showing the typical metach-romasia of cartilage. Chondrogenic differentiation was demonstrated by the mucopolyssaccharide-rich extracellular matrix (Fig. 4E, G). Chondrogenic treatment resulted in specific expression of COL2A, SOX9, and aggrecan, all of which were undetected or had basal expression in noninduced cells (Fig. 5D).
Myogenesis
After 10 days in myogenic medium, cASCs formed multinucleated structures. Controls maintained only with GM did not contain any multinucleated structures. To confirm the myogenic differentiation, the expression of myosin by immunofluorescence was assessed after 45 days (Fig. 4H). The specificity of this assay was corroborated by the absence of staining in cASCs. Expression of myogenin, dystrophin, and MyoD was observed only in induced cells, with no basal expression in control cells (Fig. 5A).
GFP Transduction of cASCs
Transgene expression was examined by flow cytometry 72 h posttransduction. About 75% of cells were GFP positive and GFP expression did not decline during culture passages (Fig. 6). To evaluate if GFP interfered with the multipotent capacity of cASCs, both GFP-positive and -negative cells at successive passages were analyzed by flow cytometry and multilineage differentiation, revealing no influence of GFP on the cellular response to inductive media (Fig. 4).

GFP-positive cASCs. (A) GFP-positive cASCs at passage 8. (B) Percentage of positively GFP cells as analyzed by flow cytometry.
Discussion
Zuk et al. (35) were the first to describe the isolation and characterization of human stem cells derived from adipose tissue. These cells were able to differentiate into adipogenic, osteogenic, chondrogenic, and myogenic lineages when exposed to inductive media.
Human ASCs are usually obtained from fat tissue that is discarded after liposuction cosmetic surgery (35). Adipose tissue can be harvested in large quantities with minimal morbidity in several regions of the body and, on average, 100 ml of human adipose tissue yields about 1 × 106 stem cells (14). In dogs, the adipose tissue can be collected by a simple adapted liposuction surgery, through biopsies or in routine veterinary surgery procedures because we could isolate cASCs from just 100 μl of adipose tissue.
In the present study we show the isolation and characterization of the canine adipose-derived stromal cell (cASC) population. While this manuscript was in preparation, Neupane et al. (17) published an article reporting the isolation of canine adipose stem cells. However, their article lacked important characteristics of the isolated cell population such as the immunophenotype, the myogenic and chondrogenic potential, and karyotype analysis at late passages. These characteristics are important for veterinary cell therapy and preclinical studies.
Our results show that cASCs can be harvested by a rapid process, an important step towards preclinical studies of cell therapy. Using this methodology, we were able to harvest cells from 10 canine subcutaneous fat samples (2 from liposuction and 8 from biopsy) with a 100% rate of success. Other groups reported successful isolation of adipose stem cells from other mammals, such as rabbit, mice, horse, and pig (27,29,32,33).
We observed that the plastic adherent cells obtained after isolation can be expanded in vitro, reaching numbers that would be sufficient for a therapeutic assay, without any numeric chromosome alteration. In addition, cASCs can be stored frozen in liquid nitrogen without cell death.
During the first days in culture, endothelial cell populations were found in the plates; however, these cells were not seen after passage 4. These data are in accordance with Rodriguez et al. (21), where the isolation of hASCs by adherence properties was reported. At passage 4, cASCs show a fibroblast-like morphology commonly found in mesenchymal stem cell (MSCs). The analysis of the cell surface markers showed that the cASCs cell population expresses the known immnophenotype of MSCs (35). At passage 4 the majority of the cells are positive for CD29, CD44, and CD90. cASCs do not express the hematopoietic marker CD45, but 10% of the cells are CD34 positive. Traktuev et al. (28) described that the population of CD34-positive cells that are found in human adipose stromal-vascular fraction are reside in a periendothelial location. The authors showed that these cells are CD31 negative. This result is in accordance with our finding with cASCs, because we found CD34-positive cells but not CD31-positive cells. This adherent cASCs cell population (CD34+/CD31-) may also interact with endothelial cells at the perivascular niche. However, further studies will be essential to identify the localization of these cells at the canine adipose tissue. In order to evaluate the MSC property of cASCs, we subjected them to differentiation studies.
MSCs are defined by their ability to self-renew and their capacity to generate committed cells in vitro and in vivo. Human ACSs can be induced to differentiate along the adipogenic, chondrogenic, and osteogenic lineages using specific culture medium (32). Even plated into scaffolds they survive long-term culture and could be terminally differentiated into adipocytes and osteo-blasts (18). In all 3 of our 10 lineages obtained we demonstrated the multipotency and plasticity of cASCs by their differentiation in adipogenic, chondrogenic, myogenic, and osteogenic lineages. The differentiation was confirmed by the appearance of lipid vacuoles, muco-polysaccharide-rich extracellular matrix, myosin labeling, and calcium deposits, respectively. Although we observed a morphological change in cells submitted to adipogenic differentiation, we found poor lipid vacuoles in cASCs when compared to hASCs submitted to the same conditions.
The stable GFP expression after cASC transduction represents a great advantage in preclinical studies. GFP-positive cells could be screened though fluorescence microscopic imaging and flow cytometry, and no significant decline of GFP expression during cASC differentiation was found. This property allows their potential use for in vitro (29) and in vivo assays (3), where cell marking with GFP was used to track stem cells seeded on a scaffold, fusing to other cells and engraftment into different tissues.
Successful canine stem cell transplantation was reported by Sampaolesi et al. (24). According to these authors, the intra-arterial delivery of canine mesoangi-oblasts (vessel-associated stem cells) resulted in an extensive recovery of muscle morphology and function in golden retriever muscular dystrophy (GRMD) dogs. Because ASCs are much easier to be obtained and can be injected without immunosuppression (11,31), they might represent a promising alternative to canine mesoangi-oblasts for GRMD stem cell therapeutic trials.
In short, results from the present study demonstrate that adherent cells isolated from canine adipose tissue can be defined as multipotent MSC with the ability to differentiate into at least four mesodermal lineages. Mouse models of human diseases have been extensively used in preclinical studies. However, mouse models have several limitations when extrapolating to humans: the small size of the mouse limits the proliferative demand placed on transplanted tissue and the short life span of the mouse also prevents long-term follow-up. Large-animal models, such as the dog, more faithfully mimic human pathologies. There are many canine animal models of human genetic diseases that are well characterized, making them ideal for in vivo studies. The characterization of cASCs represents a valuable tool for in vitro and in vivo preclinical evaluation and for the screening of therapeutic drugs. These findings also have potential relevance to future canine veterinary tissue engineering and regenerative veterinary medical therapies.
Footnotes
Acknowledgments
We gratefully acknowledge our colleagues Tatiana Jazedje, Marcos Valadares, Maria Denise Carvalho, Amanda Assoni, Camila Almeida, Mayra Pellati, Bruno Lima, Heloisa Caetano, Constancia Urbani, Dr. Mariz Vainzof, and Dr. Maria Rita Passos-Bueno for helpful suggestions as well as the earlier support of the veterinarians. This research was supported by FAPESP-CEPID (Fundação de Amparo à Pesquisa do Estado de São Paulo-Centro de Pesquisa, Inovaçào e Difusão), CNPq (Conselho Nacional de De-senvolvimento Científico e Tecnológico), and INCT (Instituto Nacional de Ciência e Tecnologia: células-tronco em doenças genéticas).