
Abstract
Stem cells from dental tissues have been isolated and established for tooth regenerative applications. However, basic characterization on their biological properties still needs to be investigated before employing them for effective clinical trials. In this study, we compared the telomere length, relative telomerase activity (RTA), and relative reverse transcriptase activity (RRA) as well as the surface antigen profile and mesenchymal differentiation ability in human dental papilla stem cells (DPaSCs), dental pulp stem cells (DPuSCs), and dental follicle stem cells (DFSCs) with mesenchymal stem cells (MSCs) derived from bone marrow. Dental stem cells (DSCs) were strongly positive for cell surface markers, such as CD44 and CD90. However, slightly lower expression of CD105 was observed in DPaSCs and DPuSCs compared to DFSCs and MSCs. Following specific induction, DPaSCs, DFSCs, and MSCs were successfully differentiated into adipocytes and osteocytes. However, DPuSCS, in particular, were able to differentiate into adipocytes but failed to induce into osteogenic differentiation. Further, all DSCs, MSCs, and MRC-5 fibroblasts as control were investigated for telomere length by nonradioactive chemiluminescent assay, RTA by relative-quantitative telomerase repeat amplification protocol (RQ-TRAP), and RRA by PCR-based assay. Mean telomere lengths in DPaSCs, DPuSCs, DFSCs, and MSCs was ~11 kb, and the values did not differ significantly (p < 0.05) among the cells analyzed. RTA levels in DPaSCs were significantly (p < 0.05) higher than in MSCs, DPuSCs, DFSCs, and MRC-5 fibroblasts and among DSCs, DFSCs showed a significantly (p < 0.05) lower RTA. Moreover, RRA levels were significantly (p < 0.05) higher in DPaSCs, DPuSCs, and MSCs than in DFSCs. Based on these observations, we conclude that among DSCs, DPaSCs possessed ideal characteristics on telomere length, telomerase activity and reverse transcriptase (RTase) activity, and may serve as suitable alternative candidates for regenerative medicine.
Keywords
Introduction
Generally, mesenchymal stem cells (MSCs) derived from bone marrow are one of the most studied and clinically important populations in the adult stem cell category. Because of some shortcomings associated with the isolation and expansion of MSCs from bone marrow, attempts have been made to explore other alternate sources. In this regard, stem cells with similar features to MSCs have been identified from dental tissues as a putative stem source (11,14,20,27,40). In humans, many different types of dental stem cells (DSCs), including dental pulp stem cells (DPuSCs), stem cells from exfoliated deciduous teeth (SHED), periodontal ligament stem cells (PDLSCs), stem cells from apical papilla (SCAP), and dental follicle progenitor cells (DFPCs), have been isolated and characterized from dental tissue [reviewed in (13)]. These populations possess MSC characteristics, including the expression of cell surface markers and alkaline phosphatase (AP) activity, the capacity for self-renewal, and multilineage differentiation potential, such as osteogenic, chondrogenic, adipogenic, myogenic, and neurogenic lineages (12,20,40,46). Moreover, the non-decayed extracted tooth of patients, such as impacted third molars (wisdom teeth) offer to serve as a better source of DSCs. Even though it has been shown that the general biological characteristics of DSCs derived from different sources are common and comparable, major differences have been demonstrated in terms of expansion and differentiation potential (10,13,18,23,40). It is considered that these differences may influence the stemness and biological properties among DSCs, and demands a careful consideration before using them for therapeutic applications.
The role of telomere and telomerase as critically important biological features of normal tissue stem/progenitor cells has been recently highlighted by numerous studies. Telomeres are specialized region of repetitive DNA localized at the end of eukaryotic chromosomes with linear DNA strands. Telomeric repeats are surrounded by many telomere-specific proteins, and these complexes provide the DNA strand with protection from destruction. Each cell division in normal somatic cells results in a progressive loss of telomeric repeats by the end replication problem during DNA replication (15). Progressive telomere shortening by the loss of telomeric repeats induces cellular senescence, and it is related to cellular life span or proliferation capacity during the prolonged serial culture of cells (1,15). Furthermore, the importance of telomere length in maintaining the self-renewal capacity and lineage differentiation has been clearly demonstrated in human stem cells (7,48).
The length of telomeric repeats can be continually maintained or extended by telomerase activity, which consists of the telomeric RNA and telomerase reverse transcriptase (TERT) with several additional telomerase-associated proteins. The level of telomerase activity is normally quite high in undifferentiated mammalian embryos and embryonic stem cells (ESCs) with long telomere length, but the level is low or undetectable in most differentiated somatic cells, resulting in gradual telomere shortening and limited capacity for proliferation (17,34). However, the level of telomerase activity is also elevated in the majority of solid tumor cell lines with short telomere length (31). Thus, the high level of telomerase activity in cell lines is associated with immortality as well as the undifferentiated condition, whereas lack of telomerase activity indicates cellular aging and senescence (9). Furthermore, it has been reported that a high level of endogenous reverse transcriptase (RTase) activity, which may have a different role from TERT, is detected in the embryo and tumor cell lines (2,30). The nontelomeric RTase, RNA-dependent DNA polymerase, contributes to retrotransposon elements which move from one position in the genome to another by replacement through RNA intermediates in the eukaryotic genome (38,42). Although obvious functions of these RTase and retrotransposons are not fully understood in the eukaryotic genome, it has been showed that nontelomeric RTase plays a key role in early embryo development and tumorigenesis (2,30). Moreover, the nontelomeric RTase activity has been detected in the human adult stem cell lines in our laboratory (unpublished data).
Although the intrinsic differences have not been comprehensively understood in DSCs compared to bone marrow MSCs, the level of telomerase activity related to telomere length and RTase activity of cell line have been considered as general markers of regenerative capacity and differentiation potential for tooth tissue engineering, such as periodontal treatments and dental implantology. Therefore, the present study compared the telomere length, telomerase activity, and RTase activity of human DSCs, such as dental papilla stem cells (DPaSCs), DPuSCs, and dental follicle stem cells (DFSCs) to those of MSCs derived from bone marrow.
Materials and Methods
Chemicals and Media
All chemicals used in the present study were purchased from the Sigma Chemical Company (St. Louis, MO, USA) and media from Gibco (Invitrogen Corporation, Grand Island, NY, USA), unless otherwise specified. For all the media, the pH was adjusted to 7.4 and the osmolality to 280 mOsm/kg.
Isolation and Culture of Mesenchymal Stem Cells From Dental Tissue and Bone Marrow
Human impacted third molars with growing and not erupted status were surgically removed from nine female patients (16–18 years of age) as a part of prophylactic treatment for orthodontic reasons at the Dental Clinic of Gyeongsang National University Hospital under approved guidelines set by the GNUH IRB-2009-34 and also after obtaining the informed consents from the patients. The teeth were stored in Dulbecco's phosphate-buffered saline (D-PBS) on ice and brought to the laboratory. After cleaning the tooth with D-PBS containing 1.0% penicillin-streptomycin (10,000 IU and 10,000 μg/ ml, respectively, Pen-Strep; Gibco), dental papilla, dental pulp, and dental follicle tissues were isolated from the tooth. Dental papilla and follicle tissues were gently collected from the surface of the extracted tooth. Dental pulp tissues were collected from the pulp chamber of the dental crowns following the fracture with bone forceps into several parts. The collected three types of tissues were cleaned with D-PBS containing 1.0% penicillin-streptomycin and then minced by using a sterilized scalpel. The isolated clumps of three types of cells were digested by incubating in D-PBS containing 1 mg/ml collagenase type I at 37°C with gentle agitation for 30 min. After washing, the clumps of cells were neutralized by adding 10% fetal bovine serum (FBS), and followed by centrifugation at 500 x g for 5 min. The clumps of cells were removed by passing the suspension through a 100μm nylon cell strainer to harvest single cell suspension. The isolated cells were primarily cultured in an Advanced Dulbecco's modified Eagle medium (A-DMEM) supplemented with 10% FBS, 1.0% Pen-Strep at 37.5°C in a humidified atmosphere of 5% CO2 in air.
For the handled control, bone marrow of 2–4 ml was extracted from an iliac crest of five female patients (16–18 years of age) who had undergone bone graft procedure for teeth implantation. MSCs were isolated according to the method of Shiota et al. (39). In brief, gelatinous bone marrow was resuspended in D-PBS and dispersed mechanically and centrifuged at 400 x g for 10 min. Cells were resuspended and layered upon a Ficoll gradient (density 1.077 g/ml, Amersham Biosciences, Uppsala, Sweden) and centrifuged at 400 x g for 40 min at 25°C. The interface buffy layer was collected and washed twice with PBS. As a control, MRC-5 normal human fetal lung fibroblasts were purchased from American Type Culture Collection (Manassas, VA, USA). All types of cells were cultured in A-DMEM supplemented with 10% FBS at 37.5°C in a humidified atmosphere of 5% CO2 in air and culture medium was changed twice a week.
As mentioned above, we isolated three types of DSCs, such as DPaSCs, DPuSCs, and DFSCs, from nine patients and MSCs from bone marrow of five patients, and subsequently subjected them to primary culture. Isolated cells exhibited more or less comparable morphological and phenotypical features after initiation of the cultures. However, cells in some cultures consisted with more heterogeneous population and showed a slightly slower proliferation rate. In contrast, the remaining cultures had more homogeneous cells with relatively uniform morphology, and, more importantly, the cells reached near confluence state earlier than others. Based on the characteristics, such as initiation of attachment, morphology, and proliferation ability, we therefore selected three cultures from each cell type, including DPaSCs, DPuSCs, DFSCs, and MSCs from bone marrow for all experiments. Selected cells were regrown by harvesting with 0.25% (w/v) trypsin-EDTA solution, and immediately analyzed or frozen at −80°C for further analysis.
Confirmative Analysis of MSCs From Dental Tissue and Bone Marrow
The isolated cells were identified with their cell surface antigen markers using a flow cytometer by following a previously described method (21) with minor modifications. The antibodies used were CD44, CD90, and CD105 (Becton Dickinson, Seoul, Korea). Briefly, the cultured DPaSC, DPuSCs, DFSCs, and MSCs at passage 3 were collected by treating with 0.25% (w/v) trypsin-EDTA, and the cells were incubated with a fluorescein isothiocyanate (FITC)-conjugated primary antibodies on ice for 1 h. Cells were then analyzed using a flow cytometer (Becton Dickinson). At least 1 × 104 cells were analyzed for each experiment.
For the evaluation of in vitro lineage differentiation, DPaSCs, SPuSCs, DFSCc, and MSCs at passage 3 were seeded at the density of 1 × 103 cell/cm2 in A-DMEM supplemented with 10% FBS under the conducive conditions for adipogenic and osteogenic differentiation by following previously published protocols (32,45) with minor modifications. Briefly, adipogenic differentiation was induced with 1 μM dexamethasone, 10 μM insulin, and 200 μM indomethacin for 4 weeks. For the evaluation of intracellular accumulation of lipids, cells were incubated for 10 min with oil red O solution. Osteogenic differentiation was induced with 0.1 μM dexamethasone, 50 μM ascorbate-2-phosphate, and 10 mM β-glycerol phosphate for 4 weeks. To confirm the calcium accumulation, differentiated cells were fixed in 3.7% formaldehyde, followed by Alizarin red staining for 30 min and von Kossa staining with 5% AgNO3 in light for 1 h at room temperature.
Analysis of Telomere Length by Nonradioactive Chemiluminescent Assay
To determine the telomere length by nonradioactive chemiluminescent assay, TeloTAGGG telomere restriction fragment (TRF) length assay kit from Roche Applied Science (Indianapolis, IN, USA) was used as per the manufacturer's instructions. Briefly, genomic DNA of the cells at passage 3 was extracted using the Qiagen DNeasy Blood and Tissue kit (Qiagen, Seoul, Korea) following the manufacturer's instructions. A total of 1.0 μg genomic DNA was digested with a mixture of HinfI and RsaI restriction enzymes at 37°C. After being separated digested DNA in 0.8% agarose gel, the samples were depurinated, denaturated, and neutralized, followed by Southern transfer onto a positively charged nylon membrane (Roche, Indianapolis, IN, USA). The membrane was hybridized with a digoxigenin (DIG)-labeled telomere probe at 42°C for 3 h. The hybridized membrane was washed in a high stringency buffer and incubated with Anti-DIG-AP solution. After the final wash, AP substrate was applied and exposed on X-ray film for 20 min at 25°C. The exposed X-ray film was scanned, and then mean length of telomere in the acquired images was calculated and analyzed using Gelviewer image processing software (Innogene, Seoul, Korea).
Analysis of Telomerase Activity by Relative-Quantitative Telomerase Repeat Amplification Protocol (RQ-TRAP)
The RQ-TRAP assay was modified from a conventional TRAP assay for its use on the LightCycler 3.0 (Roche) as previously described by Betts et al. (3). Briefly, after harvesting 1 × 105 cells at passage 3, samples were either immediately frozen at −80°C for future analysis or lysed. Cells at a concentration of 250 cells/ μl were lysed in 0.5% (v/v) 3-[(3-cholamidopropyl)di-methylammonio]propanesulfonic acid (CHAPS) lysis buffer (pH 7.5) supplemented with 10 mM Tris-HCl, 1 mM MgCl2, 1 mM EGTA, 0.1 mM benzamidine, 5 mM 2-mercaptoethanol, and 10% glycerol for 30 min on ice. Following lysis, samples were centrifuged for 20 min at 12,000 x g at 4°C to remove cell debris. After being measured for protein concentration by spectrophotometer (Mecasys, Seoul, Korea), 5 μg of total protein was analyzed by RQ-TRAP. Each run included measurements of telomerase-positive control 293T (Chemicon, Billerica, MA, USA) samples and telomerase-negative control 293T samples inactivated by incubation for 10 min at 85°C. The RQ-TRAP was optimized using the PCR reagent LightCycler FastStart DNA Master SYBR Green 1 (Roche) according to the manufacturer's protocol, containing 2.5 mM MgCl2, 0.02 μg of primer TS (5′-AATCCGTCGGAGCAGAGTT-3′), 0.04 μg of primer ACX (5′-GCGCGGCTTACCCTTACCCTTACCCTAA CC-3′), and adjusted to 20 μl of total volume using sterile H2O. The assay run included 30-min incubation at 30°C, followed by 10-min incubation at 94°C, and 40 cycles of PCR at 94°C for 30 s and 60°C for 90 s. All samples were quantified using the LightCycler Quantification Software's (Roche) second derivative method of crossing point determination, and relative telomerase activity (RTA) was calculated to a ratio based on the level of telomerase activity in the MSCs at passage 3.
Analysis of Reverse Transcriptase Activity
The relative reverse transcriptase activity (RRA) was analyzed with cell lysates containing the T enzyme by PCR-based assay as previously described by Mangiacasale et al. (24) with minor modifications. Briefly, the total protein of cells at passage 3 was extracted with CHAPS lysis buffer. Reverse transcriptase (RT) reactions contained cell lysate aliquots corresponding to 15 μg of total protein, 10 ng of purified bacteriophage MS2 RNA (Roche), 2 μl of reverse transcription buffer (Qiagen), 1 mM of four nucleotide triphosphate mix (Invitrogen, Seoul, Korea), 2 U of RNaseOUT (Invitrogen), and 30 pmol of MS2 reverse primer (see below) in a final volume of 20 μl. Reaction mixtures were incubated at 42°C for 1 h followed by 5 min at 85°C to inactivate the RNase. For removing the spared MS2 RNA after cDNA synthesis, 2 U of E. coli RNase H (Invitrogen) was added to each sample and further incubated at 37°C for 20 min in a PTC-200 Peltier thermal cycler (MJ Research, Ramsey, MN, USA). Positive control reactions were added with 1 U of omniscript reverse transcriptase (Qiagen) and negative control reactions were added with DEPC water instead of cell lysate, respectively. After cDNA synthesis, 4 μl of each reaction was amplified with 10 pmol each of forward (5′-ACCTCCTCTCTGG CTACCGA-3′), reverse (5′-ACAGGCAGCCCGATC TATTT-3′), and PCR premix kit (Intron, Daejeon, Korea), adjusted to a total volume of 20 μl using H2O. PCR conditions were as follows: 10-min incubation at 94°C, and 40 cycles of PCR at 94°C for 30 s and 60°C for 90 s. The PCR amplification product was a 297-bp DNA fragment spanning positions 2225–2521 at the 5′ end of the MS2 RNA. The PCR products were confirmed through 1.0% agarose gel electrophoresis. The intensity of PCR products by RT activity in the acquired images was calculated analyzed using Gelviewer image processing software (Innogene). RTase activity in MSCs at passage was considered as 100% for comparison with other cell lines.
Statistical Analysis
Differences among the treatments were analyzed by using one-way analysis of variance (ANOVA) by SPSS 15.0 (Chicago, IL, USA). Data were expressed as mean ± SEM. Comparisons of mean values among the treatment were analyzed using a Tukey's multiple comparisons test. A five percent probability (p < 0.05) was used as the level of significance.
Results
Analysis of Cell Surface Markers in DSCs and MSCs
The results of immunophenotypic analysis of cell surface antigens analyzed by flow cytometry in DPaSCs, DPuSCs, DFSCs, and MSCs at passage 3 are presented in Figure 1. Expression levels of CD44 and CD90 were significantly (p < 0.05) higher in DPaSCs, DPuSCs, and DFSCs than those of MSCs (98.6 ± 0.8 and 98.3 ± 0.3, 95.0 ± 1.5 and 94.6 ± 1.5, 96.0 ± 1.0 and 95.2 ± 1.8 vs. 86.0 ± 0.8 and 80.0 ± 1.7, respectively). But the expression levels were not significantly (p < 0.05) different among DPaSCs, DPuSCs, and DFSCs, as shown in Table 1. However, the expression levels of CD105 in DPaSCs and DPuSCs were significantly (p < 0.05) lower than those in DFSCs and MSCs (48.3 ± 1.8 and 47.0 ± 0.6 vs. 84.6 ± 1.5 and 79.3 ± 1.2, respectively).
Expression Levels of Cell Surface Markers in Dental Papilla Stem Cells (DPaSCs), Dental Pulp Stem Cells (DPuSCs), Dental Follicle Stem Cells (DFSCs), and Mesenchymal Stem Cells (MSCs) Derived From Bone Marrow

Values represent the mean percentage of expression level of specific markers assessed in different cells stained against the respective antibodies. Three replicates.
Values in columns differ significantly (p < 0.05).

Analysis of cell surface markers by flow cytometry in dental papilla stem cells (DPaSCs), dental pulp stem cells (DPuSCs), dental follicle stem cells (DFSCs), and mesenchymal stem cells (MSCs) at passage 3 in three replicates. Each cell line derived from DPaSC, DPuSC, DFSC, and MSC samples were labeled with antibodies against CD44, CD90, and CD105, and analyzed by flow cytometry in triplicate with 10,000 cells from every sample. Open and filled histograms indicate isotype-matched controls and fluorescence intensity of each cell surface marker, respectively. Only representative examples indicating specific marker expression levels are depicted.
In Vitro Differentiation of DSCs and MSCs
Following 4 weeks of induction in adipogenic- and osteogenic-specific media, differentiated cells were analyzed for the formation of lipid clusters and accumulation of mineralized nodules of calcium, respectively (Fig. 2). Intercellular formation of neutral lipid droplets stained with oil red O solution was clearly exhibited in DPaSCs, DPuSCs, DFSCs, and MSCs (Fig. 2A, D, G, J). Accumulation of calcium mineralized nodules as indicated by von Kossa (Fig. 2B, H, K) and Alizarin red staining (Fig. 2C, I, L) was observed in adherent cell cultures of DPaSCs, DFSCs, and MSCs, However, osteogenic differentiation could not be detected in DPuSCs after 4 weeks of culture in specific medium (Fig. 2E, F).
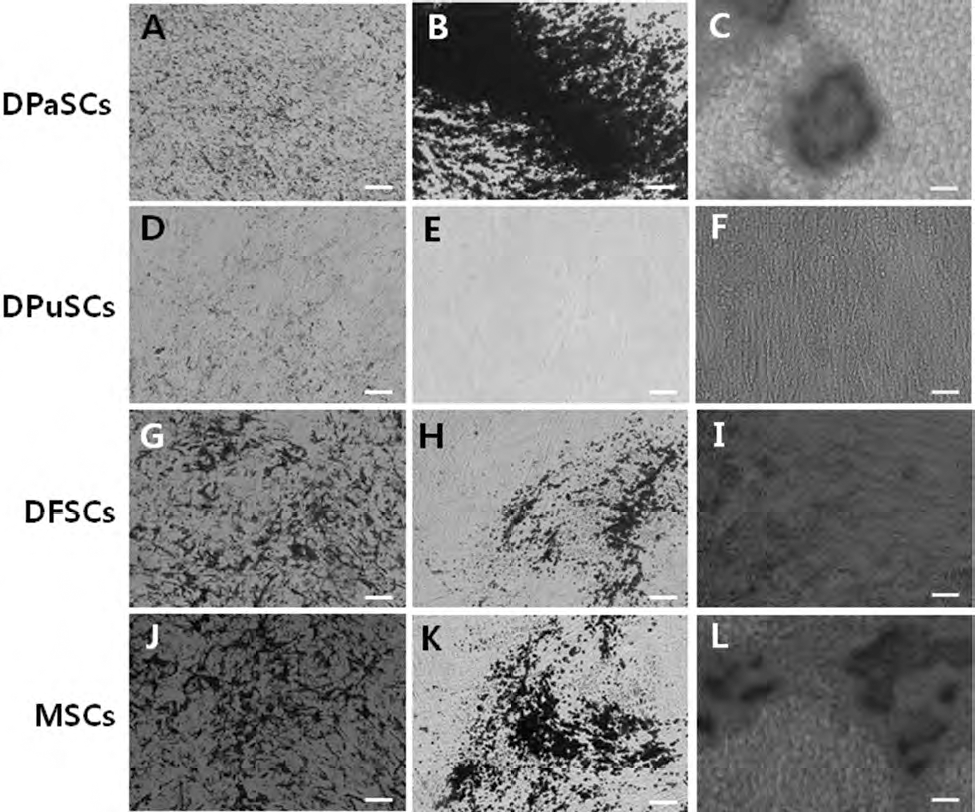
Adipogenic and osteogenic differentiation in DPaSC, DPuSC, DFSC, and MSC samples at 4 weeks after induction of differentiation. Each cell line derived from DPaSC, DPuSC, DFSC, and MSC samples was used for differentiation induction. Intracellular accumulation of lipids stained as dark spots by oil red O solution staining in the DPaSCs (A), DPuSCs (D), DFSCs (G), and MSCs (J). DPaSCs, DFSCs, and MSCs showed their ability to differentiate into osteocytes through the accumulation of calcium, indicated by von Kossa (B, H, and K, respectively) and Alizarin red (C, I, and L, respectively) staining. Osteogenic differentiation was not observed in DuPSCs by both von Kossa (E) and Alizarin red (F) staining. Scale bars: 50 μm.
Telomere Length in DSCs and MSCs
The range of telomere length in DPaSC, DPuSC, DFSC, MSC, and MRC-5 fibroblasts varied from 8.77 to 14.96, 9.36 to 12.05, 10.28 to 12.80, 7.50 to 16.12, and 4.02 to 10.67 kb, respectively (Fig. 3A). Furthermore, the mean of telomere length in the spot that showed the highest density was 11.9 ± 1.20, 10.6 ± 0.65, 11.2 ± 0.67, 11.3 ± 1.46, and 7.0 ± 1.12 kb in DPaSC, DPuSC, DFSC, MSC, and MRC-5 fibroblasts, respectively (Fig. 3B). Telomere length was not significantly (p < 0.05) different among DPaSCs, DPuSCs, DFSCs, and MSCs. However, the telomere length in these cells was significantly (p < 0.05) higher than that of MRC-5 fibroblasts (Fig. 3B).
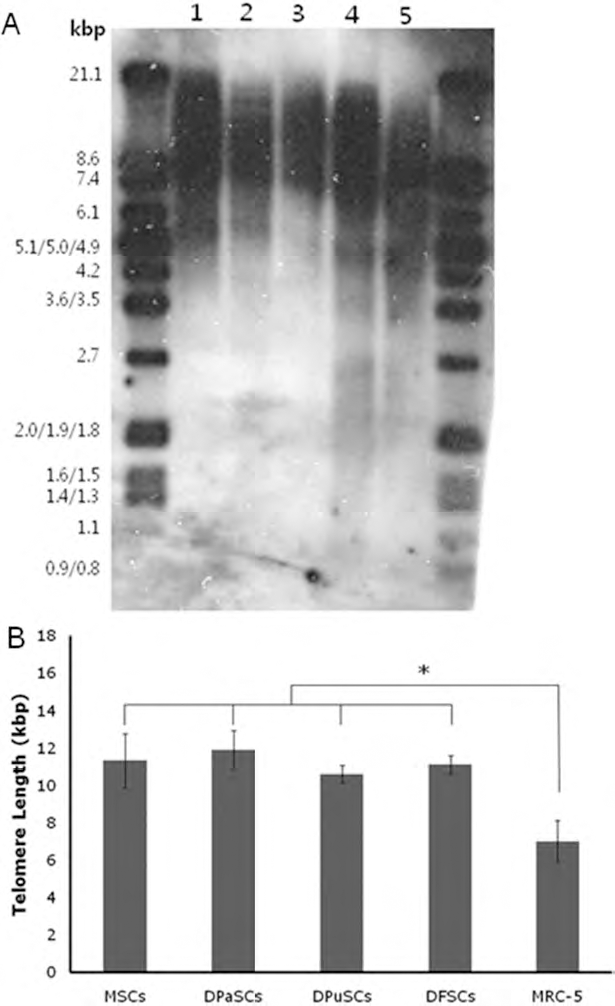
Telomere length in MSC, DPaSC, DPuSC, DFSC, and MRC-5 fibroblasts (from normal human fetal lung as control). (A) The range of telomere restriction fragment length by non-radioactive chemiluminescent assay. Lane 1: DPaSC; lane 2: DPuSC; lane 3: DFSC; lane 4: MSC; lane 5: MRC-5 fibroblasts. (B) Mean telomere lengths in DPaSC, DPuSC, DFSC, and MSC samples. Values represent the percentage mean ± SEM of telomere lengths analyzed for all samples. ∗p < 0.05.
Telomerase Activity in DSCs and MSCs
For comparison with other cell lines, RTA in MSCs at passage 3 was considered as 100%. The values observed for RTA in MSC, DPaSC, DPuSC, DFSC, and MRC-5 fibroblasts were 100%, 121%, 101%, 62%, and 36%, respectively (Fig. 4). RTA in DPaSCs was significantly (p < 0.05) higher than in MSC, DPuSC, DFSC, and MRC-5 fibroblasts, and among DSCs, DFSCs recorded a significantly (p < 0.05) lower RTA.

Relative telomerase activity (RTA) by relative-quantitative telomerase repeat amplification protocol (RQ-TRAP) assay in MSC, DPaSC, DPuSC, DFSC, and MRC-5 fibroblasts at passage 3. Values indicated the mean telomease activity (mean ± SEM) of five replicates in DPaSC, DPuSC, DFSC, and MSC samples and the telomerase activity in MSCs was considered as 100% for comparison with other cell lines. ∗p < 0.05.
RTase Activity in DSCs and MSCs
To determine RRA, cell lysates extracted from MSC, DPaSC, DPuSC, DFSC, and MRC-5 fibroblasts at passage 3 were subjected to PCR-based RTase assay and the results are represented in Figure 5A. RRA in MSCs was considered as 100% for comparison with other cell lines. RRA was 100%, 110%, 90%, 63%, and 0% in MSC, DPaSC, DPuSC, DFSC, and MRC-5 fibroblasts, respectively (Fig. 5B). RRA in MSCs, DPaSCs, and DPuSCs was significantly (p < 0.05) higher than in DFSCs. However, RRA was not detected in MRC-5 fibroblasts and, as expected, in negative control reactions. The level of RRA in MSCs was about one third of that of positive control reactions.

Relative reverse transcriptase activity (RRA) in MSC, DPaSC, DPuSC, DFSC, and MRC-5 fibroblasts at passage 3. (A) PCR-based reverse transcriptase (RT) activity assay using MS2 RNA and lysates. Lane 1: MSC; lane 2: DPaSC; lane 3: DPuSC; lane 4: DFSC; lane 5, MRC-5 fibroblasts; lane 6: positive control reaction with RT enzyme for RT-PCR method; lane 7: negative control without lysates; lane 8: negative control without cDNA. (B) Mean RRA in MSC, DPaSC, DPuSC, DFSC, and MRC-5 fibroblasts. Values indicated the mean RTase activity (mean ± SEM) of five replicates in DPaSC, DPuSC, DFSC, and MSC samples and the RTase activity in MSCs was considered as 100% for comparison with other cell lines. ∗p < 0.05. ND, not detected.
Discussion
In the present study, we isolated a population of stem cells from the dental tissues of third molars, and compared their expression levels of CD markers, adipogenic and osteogenic potential, telomere length, telomerase activity, and RTase activity with MSCs from bone marrow. Our results demonstrated that DPaSCs derived from dental tissue possess similar biological features compared to the bone marrow MSCs, whereas the characteristics of DFSCs and DPuSCs were demonstrated to be slightly different.
Expression of CD markers and lineage differentiation ability have been evaluated for verification of stemness in the stem cell lines derived from dental tissues (12,16,20,40). Expression or nonexpression of CD markers, such as CD105+, CD73+, CD90+, CD14-, or CD11b-, CD34-, CD45-, CD19-, or CD79α-, and human leukocyte antigen (HLA)-DR- in cells supports their mesenchymal origin (5). Since there is no single specific marker available to identify MSCs, combinations of markers have been employed by different groups. In this study, expression levels of CD44 and CD90 in DSCs were higher than in MSCs, but were found to be similar among DSCs. Our observations are in accordance with earlier reports showing the higher expression of these specific markers in various DSCs (16,20,23,28). However, we observed the expression level of CD105 at slightly lower levels in DPaSCs and DPuSCs than in DFSCs and MSCs. Interestingly, total pulp cells from human dental tissue exhibited a very low (4.4%) level of CD105 expression (28). Further, in another study, only 9.23% of stem cells from root apical papilla revealed the expression of CD105 (40). CD105 is expressed mainly in endothelial cells (6), and the fact that MSCs share some common features with endothelial cells may present a highly variable profile of this cell surface antigen. Furthermore, the presence of a specific, distinct antigen that is identified on the cell surface of DFSCs at higher levels, and the same antigen exists at slightly lower levels in DPaSCs and DPuSCs suggests that the expression of recognized epitope may have been developmentally regulated in the dental tissues. In addition, variations in expression may also be due to differences in sample origin, development stage, culture techniques, and media composition or differences in the donor age and type from which the DSCs were obtained and used for immunophenotypic analysis. Results of protein markers expression suggests that DSCs in the present study are comparable to MSCs and may have originated from similar precursor cells. But their exact identity is not conclusively proven due to lack of consistent data. Hence, these results indicate that dental tissues may typically contain various cell populations including multipotent stem, progenitor, precursor cells and terminally differentiated cells.
Relevance of the differences observed for marker expression in the present study has been inconclusive and, hence, the results of differentiation towards mesenchymal lineages would be more appropriate to characterize DSCs isolated from various tissues. Similar to MSCs, DPaSCs and DFSCs demonstrated the capacity to undergo adipogenic and osteogenic differentiation following in vitro induction. The findings of the present study supported the previous results (16,18,20), but the differentiation abilities were varied among the three cell types from dental tissues. DPuSCs were more effective in forming adipocytes, but could not differentiate into osteocytes when compared to DPaSCs and DFSCs. More recently, DPuSCs were also found to undergo weaker adipogenic differentiation in vitro in comparison to bone marrow MSCs (41). Furthermore, osteogenic differentiation was achieved in each stem cell line derived from dental tissues, including dental pulp, dental follicle, and periodontal ligament, but adipose differentiation of the cell lines could not be established (23). The reasons for these conflicting results from others and our study are not fully deciphered. Basically, odontoblastic differentiation potential has been shown as an important feature of pulp cells (13). DPuSCs can easily induce differentiation into odontoblast-like cells to form a unique dentin extracellular matrix in the pulp chamber. This property may indicate their intrinsic tendency of differentiation towards odontoblasts and less sensitivity for the formation of osteocytes and adipocytes. Supporting these observations, human DPuSCs formed a dentin-like tissue that surrounded a vascularized, pulp-like tissue when transplanted into immunocompromised mice (11). Moreover, dental pulp harbors greatest numbers of fibroblasts in their varying progressive levels of maturation. Like MSCs, they are also spindle shaped and the presence of matured fibroblasts in large numbers in our cultures may have hindered their capability to undergo extracellular mineralization. Further, the results also show that the dental cell populations may contain cells of varying inherent differentiation ability or may contain cells of different origin, such as mesodermal and ectodermal (13). Hence, there are significant differences in stem cell properties and, more importantly, the difference in donor type and tissue origin of DSCs may determine their multipotency and functional characteristics. Taken together, we summarize that general biological characteristics of DSCs obtained from different sources are relatively similar, but differences may exist in terms of surface antigen profile and differentiation potential.
The status of senescence or aging in mammalian cells is triggered by various factors of cellular damage and, among them, DNA damages by telomere shortening is one of the most important factors (35). It has been shown that the stability of DNA is increased in the cells with longer telomere length than those of shorter telomere length and telomere length is decreased during in vitro culture (26). Moreover, the telomerase activity, responsible for the maintenance or extension of telomere length, was increased in most solid tumor cells with short telomere length, and these cells attain immortal status by the maintenance of telomere length (36).
It has been reported that human dental papilla cells, dental pulp, and periodontal ligament cells could be immortalized by transfection of TERT (19). Thus, telomerase activity as well as telomere length may have an effect on the cellular senescence and life span. Undifferentiated ESCs generally showed no loss of telomerase activity and proliferative capacity; however, telomerase activity was gradually decreased in the differentiating or differentiated ESCs (29). On the other hand, the elevated activity of telomerase in the embryonic and adult stem cells has been related to their self-renewal ability, which sustains cell division and provides stability to the chromosomes (22). In these points of view, the long telomere length and elevated telomerase activity observed in DPaSCs than in DPuSCs and DFSCs indicate the sustained capacity of cell division with undifferentiated status. It has been showed earlier that telomere length in stem cells derived from the dental pulp of adult chimpanzee remains between 10 and 20 kb (4). In the present study, telomere length in DSCs was ~11 kb and it was found similar among DPaSCs, DPuSCs, DFSCs, and MSCs. However, the erosion of telomere repeats may take place faster in the DPuSCs and DFSCs with low level of telomerase activity than that in DPaSCs with high level of telomerase activity, resulting in the decrease of proliferative potential. Similar observations have been reported with higher level of telomerase activity in SCAP than that of DPuSCs in swine (40). Further, telomerase activity in DPuSCs isolated from natal teeth was higher than MSCs from bone marrow of 4–7-year-old patients. This variation was probably observed due to the age difference between DPuSCs and MSCs. Moreover, the levels of telomerase activity in the bone marrow MSCs have been different between the isolated cell lines and assay method for the measurement of telomerase activity. Telomerase activity in human MSCs analyzed with TRAP-ELISA method was detected at very low levels by exhibiting a rare group of MSCs subpopulations (49). The level of telomerase activity was undetected in the somatic cells such as fibroblasts; however, the telomerase activity was detected in the adult fibroblasts and this might be due to the greater sensitivity of real time RQ-TRAP method (17).
The biological characterization on RTase activity in adult stem cells has not been reported previously. Here we detected the higher levels of RTase activity in DSCs as well as MSCs. RTase generally transcribes single-stranded RNA into double-stranded DNA. Telomerase is also a kind of RTase; however, the function of RTase is dissimilar to telomerase in the cells (38). Even though the function of nontelomeric RTase has not been fully demonstrated, nontelomeric RTase coding genes were expressed in the early embryos (33), germ cells (8), and embryonal carcinoma cells (25) at higher levels. Inhibition of RTase by microinjection of an anti-RTase antibody arrested the preimplantation stages in the mouse embryo (2). The presence of endogenous RTase activities in swine and human spermatozoa has been regarded as an inherent characteristic of male germ cells of mammalian species (38). Furthermore, earlier findings indicated the possible role of RNA-dependent or RTase-dependent flow of epigenetic information carried by sperm cells (8,37). In another study, RTase inhibition by transient silencing of RT-encoding LINE-1 (long interspersed nuclear elements) by RNA interference significantly reduced the tumor progression and cell proliferation, and induced the cellular differentiation in tumor cell lines (30). Propagation of the reverse transcribed structures has been supported by the presence of LINE-1 encoded RTase activity in early embryos (2) and tumor cells (30). Based on the collective evidences of previous studies [reviewed in (42)], we presume that RTase activity existing in DSCs and MSCs probably has an important role in cellular proliferation and differentiation. Recently, it has been demonstrated that somatic fibroblasts and multipotent cells are capable of acquiring the pluripotent state similar to ESCs by gene transfection of master transcriptional regulators, such as Octamer-binding transcription factor 4 (Oct4), sex determining region Y-box 2 (Sox2), c-Myc, and Kruppel-like factor 4 (Klf4) (43,44,47). The gene transfection has been typically achieved through viral vectors, such as retroviruses or lentiviruses, which produce DNA from its RNA genome with RTase activity in a host cell, implying the incorporation of Oct4, Sox2, c-Myc, and Klf4 genes into the double-stranded DNA of fibroblasts by RTase (38). Increased evidence strongly suggests that the function of RTase is probably concerned with highly proliferating and undifferentiated status of stem cells.
In conclusion, the present study clearly showed that among three types of DSCs, DPaSCs express the CD markers of multipotent stem cells with adipogenic and osteogenic differentiation capacity, and also maintain a long telomere length with higher level of telomerase activity, probably providing a large number of stem cells with increased proliferative potential. Besides, RTase activity, which seems to be related with cellular differentiation, was also detected at higher levels in DPaSCs. Thus, the results suggested that DPaSCs might provide a putative source of stem cells for tooth regeneration and repair as well as wide range of applications in regenerative medicine of humans. However, the roles of RTase on cellular differentiation need to be investigated further in stem cells.
Footnotes
Acknowledgments
This study was supported by grants from BioGreen 21, 20070301034041 and 200908FHT010204005, Rural Development Administration, Republic of Korea. The authors declare no conflict of financial interest.