
Abstract
In this study we describe the derivation and immunological characterization of a primary epithelial cell type from the human umbilical cord membrane. These cord lining epithelial cells (CLECs) expressed and/or secreted isoforms of the nonclassical human leukocyte antigen class I (HLA-1b) glycoproteins, HLA-G and E. Conditioned media from CLECs inhibited mitogen-stimulated T-lymphocyte responses, and in a mixed leukocyte reaction (MLR) assay, cocultured CLECs inhibited allogeneic responses with a concomitant reduction in proinflammatory cytokines. Using a transwell coculture system, it was demonstrated that these immunoregulatory effects were mediated by soluble factors secreted by CLECs, in a dose-dependent manner. Functional studies using HLA-G blocking antibody showed that the effects of CLEC-secreted products could be inhibited, thus demonstrating a significant and important role for soluble HLA-G. In vivo, we show that transplanted CLECs could be maintained for extended periods in immunocompetent mice where xenorejection rapidly destroyed primary keratinocytes, a control human epithelial cell type. Additionally, CLECs delayed the rejection of keratinocytes and extended their survival when cotransplanted, indicating an ability to protect adjacent human cell types that would otherwise be rejected if transplanted alone. We also show that CLECs transduced with a modified human proinsulin gene were transplanted intraperitoneally into streptozotocin (STZ)-induced diabetic mice, resulting in significantly lower levels of serum glucose compared to control mice. This study has characterized the immunological properties of CLECs and tested a potential therapeutic application in the treatment of a type 1 diabetes mouse model.
Introduction
The selection of a potential source of cells for cell transplantation depends on many factors. One of these factors may be the immunogenicity, which is the ability of the transplanted cells to provoke the host immune responses. In the present study, we have studied a novel cell type from the human umbilical cord lining and characterized their immunological properties that may enhance survival after transplantation.
Human umbilical cord lining-derived epithelial cells (CLECs) have recently been described (19,43) and are derived from the discarded umbilical cord after delivery of the newborn. The umbilical cord, as well as the placenta, is immunoprivileged throughout pregnancy. The mechanisms that switch off maternal immune responses and lead to a temporary tolerance of the fetal allograft are not fully understood, and are thought to be primarily mediated by the placenta. A recent study (9) has described the immunomodulatory properties of cord lining derived mesenchymal stem cells (MSCs) in comparison to MSCs derived from other tissues.
To parallel this and previous studies conducted on placental cell types, including trophoblastic and mesenchymal (1,22,24), we have analyzed CLECs for expression of two nonclassical forms of human leukocyte antigen (HLA): HLA-G and E. These forms of major histocompatibility complex (MHC) molecule are linked with placental mechanisms that modulate maternal immune rejection (16,18). HLA-G is known to have powerful immunosuppressive effects on diverse immune cell types including T cells (2), natural killer (NK) cells (34), and dendritic cells (DCs) (33). HLA-E expression is associated with a number of immune refractory tumors and regulates NK cell activity through interactions with CD94/NKG2 receptors (4).
In this study, using a coculture system and functional assays, we have demonstrated a key role for soluble HLA-G in the immunoregulation by CLECs. We have also analyzed the influence of CLECs on the cytokine milieu engendered as part of an ongoing alloresponse, which can have important consequences for the kinetics and scale of the response (5,8).
We subsequently studied the ability of CLECs to survive under conditions of xenorejection in immunocompetent Balb/c mice and recovered viable CLECs from xenotransplants after a prolonged period, which was in marked contrast to the rapid clearance of an alternative primary epithelial cell type, human keratinocytes. Finally, we transplanted CLECs transduced with the human proinsulin gene and demonstrated a reduction in serum glucose levels in mice with streptozotocin (STZ)-induced hyperglycemia in vivo.
Materials and Methods
Derivation of Primary Cell Lines
Human CLECs were isolated from the lining membrane of human umbilical cord by researchers from CellResearch Corporation, Singapore, as described previously (43) (international publication number WO 2006/019357 A1 and UK Patent GB2432166). These cells are distinct from the recently described cord lining mesenchymal cells (CLMC) morphologically (Fig. 1) and in HLA expression (9). Human umbilical cord tissues were obtained after delivery of uncomplicated pregnancies with written informed consent from donors. Dissection of umbilical cord tissue was performed to separate the umbilical cord membrane from Wharton's jelly (the matrix of umbilical cord) and other internal components. Sectioned small pieces of umbilical cord membrane were explanted to tissue culture dishes and the primary cell lines were allowed to grow out of these sections. Cultures of the CLECs were maintained in Medium 171 (Cascade Biologics, USA) supplemented with 50 μg/ml insulin-like growth factor-1, 50 μg/ml platelet-derived growth factor-BB, 5 μg/ml transforming growth factor-β1, and 5 μg/ml insulin (R&D Systems, USA).
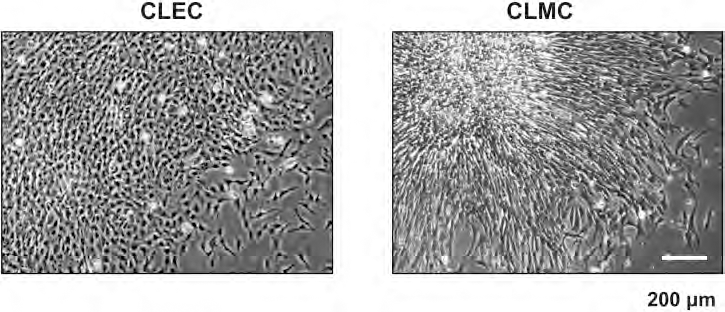
Morphology of cord lining derived cells. The cord lining-derived epithelial cell (CLEC) has a cuboidal/polygonal morphology whereas the cord lining mesenchymal cell (CLMC) is elongated and fibroblast-like.
Flow Cytometry
CLECs were harvested from culture dishes by trypsinization, immunostained, and surface marker expression analyzed with a Dako-Cytomation CyAn ADP flow cytometer. Antibodies were used against the following human antigens: HLA-A, B, C, HLA-DR, CD54 (BD Pharmingen, USA), pan-cytokeratin, CD44, and CD104 (eBioscience, USA). Permeabilization with 0.5% saponin (Sigma-Aldrich, USA) was utilized to detect intracellular expression of nonclassical MHC I molecules with the following antibodies: anti-HLA-G (clone 4H84, BD Pharmingen, USA), and anti-HLA-E (clone MEM-E/02, Abcam, UK). T cells were harvested from mixed leukocyte cultures, washed, stained, and analyzed as described above, using the following antibodies: anti-CD4 (BD Pharmingen, USA), anti-CD25 and anti-Foxp3 (Foxhead Box P3; Treg Staining Kit, eBioscience, USA). Isotype controls were used accordingly in all experiments.
Immunoblotting
Cultured cells were lysed with cell lysis buffer containing 10 mM Tris (pH 7.4), 1 mM EDTA, 150 mM sodium chloride, 0.5% Triton X-100, 0.5% Nonidet P-40, 250 μM sodium vanadate, and 1 mg/ml protease inhibitor cocktail (Boehringer-Mannheim, Germany). Soluble fractions were collected as supernatants after centrifugation of the whole cell lysate. Protein concentrations were determined by Bradford protein assay (Bio-Rad, USA). Protein samples were separated by 14% SDS-PAGE under reducing conditions and blotted onto nitrocellulose membranes. Blots were incubated with the specific antibodies against HLA-G (clone 4H84, BD Pharmingen, USA) and HLA-E (clone MEM-E/02, Abcam, UK), followed by horseradish peroxidase-conjugated rabbit anti-mouse secondary antibody (Abcam, UK). The blots were visualized with a chemiluminescence-based photoblot system (Amersham Biosciences, UK). To detect HLA-G/E in the conditioned medium, culture supernatants were concentrated using an Amicon centrifugal filtration device (Millipore, USA), quantified, and then subjected to Western blot analysis.
Reverse Transcription PCR (RT-PCR)
Total RNA was extracted from cultured cells using Qiagen RNeasy Mini kit, and quantified with Nanodrop ND-1000. Reverse transcription was done with SuperScript® III First-Strand Synthesis System (Invitrogen, USA), using 2 μg of total RNA primed with 2.5 μM oligo dT (1st BASE, Singapore) for 1 h at 50°C, with a reaction volume of 20 μl. The reaction was stopped by heating at 80°C for 5 min. PCR was done using an MJ Research PTC-100 thermal cycler, in a total volume of 50 μl with 2 μl RT product, 200 μM of each dNTP (Promega), 0.4 μM of each primer (1st BASE), 1.5 mM magnesium chloride, 10 μl 5x Green GoTaq® Flexi Buffer, and 1.25 units GoTaq® Flexi DNA polymerase (Promega, USA). PCR cycle conditions were 45 s at 95°C, 45 s at 60°C, and 45 s at 72°C, for 40 cycles, with initial denaturation at 95°C for 2 min and final extension at 72°C for 5 min. β-Actin cDNA was used as a control and amplified with 30 cycles of PCR. Primers were designed according to Refseq (NM_002127 for HLA-G, NM_005516 for HLA-E, and NM_001101.3 for human β-actin) downloaded from the NCBI sequence viewer, using online primer 3 (v. 0.4.0) (35) at http://frodo.wi.mit.edu/primer3/. Primer sequences are listed in Table 1.
Primer Sets Used in RT-PCR

Primers were designed according to Refseq (NM_002127 for HLA-G, NM_005516 for HLA-E, and NM_001101.3 for human β-actin) downloaded from the NCBI sequence viewer, using online primer 3 (v. 0.4.0) (35). RT, reverse transcription; HLA, human leukocyte antigen.
Isolation of Immune Cell Subsets
Human peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Hypaque density gradients from the peripheral blood of healthy volunteers with informed consent under an approved protocol from the National University of Singapore-Institutional Review Board (NUS-IRB). Peripheral blood monocytes were positively selected from PBMCs using anti-CD14 microbeads (Miltenyi Biotechnology, Germany). Dendritic cells (DCs) were generated by culturing the monocytes in 100 ng/ml granulocyte-macrophage colony stimulating factor (GM-CSF) and 1000 U/ml interleukin-4 (IL-4) for 7 days in DMEM containing 10% human AB serum (Cambrex, UK) (37). T lymphocytes were isolated from PBMCs using anti-CD4 or anti-CD8 microbeads (Miltenyi Biotechnology, Germany) and used directly in mixed leukocyte reactions (MLRs) or mitogen stimulation assays.
Allogeneic Mixed Leukocyte Reaction
Peripheral blood monocyte-derived DCs and T cells (CD4+ unless otherwise stated) derived from two genetically unrelated human donors were cocultured at DC/T ratio of 1:5 for 7 days, in the presence of 5 U/ml recombinant IL-2 (iDNA Biotechnology, Singapore), in DMEM with 6.67% human AB serum and 1 U/ml penicillin 1 μg/ml streptomycin, respectively (Gibco, Invitrogen, USA). Two different assays were used to quantify T-cell proliferation in the MLR.
For tritiated thymidine incorporation assays, the MLR was carried out in triplicate culture wells in 96-well microtitre plates with 2 × 105 T cells/well. Culture supernatants from CLEC cell lines were added at 100 μl/well to 50% of the final volume. Fresh culture medium without CLEC conditioning was used as control.
For carboxyfluorescein succinimidyl ester (CFSE) proliferation assays, CLECs were seeded into 24-well plates at a density of 50,000 cells/well (unless otherwise noted), 1 day before the MLR. T cells and DCs were later added directly into the culture wells (1 × 106 T cells/well) or placed into a 0.4 μm pore size cell culture insert (BD Falcon Inc, CA, USA).
Mitogen Stimulation Assay
T cells were stimulated with 2 μg/ml phytohemagglutinin (PHA) (Sigma-Aldrich, USA) for 48 h and pulsed with [3H]thymidine (GE Healthcare, UK) to determine proliferation. CLEC-conditioned culture supernatants were utilized at identical ratios (50% v/v) as those employed in the MLR assays.
[3H]Thymidine Incorporation Assay
[3H]Thymidine (GE Healthcare, UK) was added to a final concentration of 0.5 μCi/well on the seventh day of MLR or the second day of the mitogen stimulation assay. Cells were incubated for 6 h to allow [3H]thymidine incorporation into actively dividing cells. The labeled DNA was measured by harvesting the T cells onto UniFilter plates (PerkinElmer, USA), dried at 60°C for at least 1 h, and analyzed on a Beckman TopCount liquid scintillation plate reader. In the MLR experiments, DCs were pretreated with mitomycin C (15 μg/ml) (Sigma-Alldrich, USA) prior to MLR culture to ensure the measured proliferation was restricted to T cells.
CFSE Cell Proliferation Assay
T cells were stained with the membrane-permeable dye carboxyfluorescein diacetate, succinimidyl ester (CFDA-SE) (Invitrogen, USA) prior to MLR, at 2 μM for 10 min at 37°C. CFDA-SE was then processed by intracellular enzymes to become the highly fluorescent carboxyfluorescein succinimidyl ester (CFSE), which was retained inside the cell and diluted in daughter cells during division/proliferation. T cells were harvested on the seventh day of MLR, immunostained, and analyzed by flow cytometry.
HLA-G Functional Assay
The role of HLA-G in CLEC immune regulation was investigated in the allogeneic MLR using a functional grade anti-HLA-G antibody (clone 87G, Exbio, Czech). CLEC-conditioned medium was preincubated with the blocking antibody at 30 μg/ml, 37°C for 30 min. This was then added into the MLR culture resulting in a final concentration of the antibody at 15 μg/ml. CLEC-conditioned medium treated with an isotype control antibody under identical conditions served as negative control.
Multiplex Cytokine Arrays
Supernatants from CLEC and MLR cultures were collected (100 μl/well) from 24-well plates at the end of the 7-day MLR and stored at −80°C until use. Fresh medium and CLEC monoculture supernatants were also included in the assay as controls. Human cytokines were measured using multiplex cytokine assay bead suspension systems (Bio-Rad Laboratories, USA) with simultaneous detection of several human cytokines in a single sample (12). Data analysis was carried out using 5-parametric-curve fitting Bio-Plex Manager software (Bio-Rad Laboratories, USA).
Stable Transduction of CLECs and Keratinocytes
For the preparation of pseudotyped lentiviral vectors, the transfer vector plasmid, packaging plasmid pCMV ΔR8.91, and VSVG envelop protein-coding plasmid pMD.G were cotransfected into subconfluent human embryonic kidney (HEK) 293T cells by calcium phosphate precipitation, following Prof. Trono's previously described protocols (27). The viral supernatant was harvested in the respective growth medium of CLECs and keratinocytes. CLECs and keratinocytes were transduced with lentiviral vector WPT-DsRed2 and WPT-eGFP (red and green fluorescent proteins; kind gift from Prof. Didier Trono, University of Lausanne, Switzerland), respectively, with multiplicity of infection (MOI) ranging from 10 to 30 and in the presence of 8 μg/ml polybrene. The viral supernatant was removed 24 h after infection and replaced with culture medium.
For ex vivo gene therapy of diabetic mice, a lentiviral vector encoding a human proinsulin gene construct (kind gift from Prof. Ann Simpson, University of Technology, Sydney) (32) was transduced into CLECs, using the same method as described above.
Xenotransplantation
All animal experiments were done under the approval of Institutional Animal Care and Use Committee (IACUC). Surgical procedures were performed under anesthesia induced by intraperitoneal injection of 0.1 ml of a mixture consisting of equal parts of Hypnorm (10 mg/ml fluanisone and 0.315 mg/ml fentanyl citrate; Janssen Pharmaceutica, Berchem, Belgium) and Dormicum (5 mg/ml midazolam; Roche, Basel, Switzerland), diluted in two parts water. Immunocompetent Balb/c mice (8–10 weeks old) were used for assaying the survival of CLECs in vivo under conditions of acute rejection. The DsRed2 fluorescence-tagged CLEC and eGFP-tagged primary human skin keratinocytes were cultured at an identical seeding density of 2 × 105 cells/graft. The cells were transplanted as a monolayer on a porous polyethylene phthalate (PET) membrane to allow easy visualization and recovery. A 1–2-cm incision in the skin was made on the dorsal side. Three pieces of graft (6 × 105 cells in total) per mouse were inserted subcutaneously. Grafts were removed 12 and 48 h and 14 days later to assess cell survival by fluorescent microscopy and reculture of the explanted cells. To establish recultures, explanted graft was washed in PBS with 5% antibiotic antimycotic and trypsinized for 5 min at 37°C.
CLEC-Based Ex Vivo Gene Therapy
Severe combined immunodeficiency (SCID) mice (CB17/IcrHanHsd-PrkcdSCID) were treated with STZ to induce diabetes-like hyperglycemia. Freshly prepared STZ was injected intraperitoneally at a single dose of 180 mg/kg after 4 h of fasting. Mice with serum glucose levels above 14 mM (252 mg/dl) for at least 3 consecutive days were considered stably hyperglycemic. Mice of the treatment group received an intraperitoneal injection of insulin-expressing CLECs (resuspended in RPMI-1640 medium) at 1 × 107 cells per mouse in 250 μl. Equal volumes of RPMI-1640 medium alone were injected into mice of the control group. Serum glucose was monitored from the tail vein using Accu-Chek Active glucose meter (Roche Diagnostics, Switzerland), daily in the first week post-cell injection and twice a week afterwards until the end of experiment. Mice were euthanized at 3 weeks post-cell injection and the pancreata were harvested for histological analysis.
Histology
Grafts harvested from the mice were fixed, processed, embedded into paraffin blocks, and cut into 5-μm sections. For immunostaining, paraffin sections of tissues were boiled in citrate buffer for antigen retrieval and then immunostained with anti-human vimentin antibody (clone V9, Invitrogen) or anti-GFP antibody (Chemicon), followed by visualization using the Ultra-Vision LP Value Detection System HRP Polymer & AEC Chromogen kit (Thermo Fisher Scientific, USA). Hematoxylin counterstaining was used to label cell nuclei.
Statistics
Student's t-test with two-tailed p-values was used for data analysis of T-cell proliferation assays. Bonferroni correction was applied where multiple comparison was involved. Friedman test was used to analyze data from multiplex cytokine arrays. Wilcoxon signed-rank test was used as post hoc analysis, with Bonferroni correction. For ex vivo gene therapy of STZ-induced hyperglycemic mice, the statistical significance of differences between the treatment and control groups was tested using general linear model for repeated measures.
Numbers (N) are given in each figure legend. Error bars represented standard error of the mean. Statistical analysis was conducted using SPSS version 17.0.
Results
Derivation and Phenotypic Characterization of CLECs
Phenotypic analysis of CLECs by flow cytometry indicated positive surface staining for HLA-A, B, C, the classical MHC class I molecules, while staining negative for HLA-DR. They also expressed two nonclassical members of the MHC class I molecules, HLA-G and E. In addition, CLECs expressed cytokeratins and adhesion molecules that are typical of epithelial cells. These included CD44 (homing cell adhesion molecule; HCAM), CD54 (intracellular adhesion molecule-1; ICAM-1), and CD104 (integrin β4) (Fig. 2A).

Cord lining epithelial cells express the immunoregulatory molecules HLA-G and HLA-E. (A) Flow cytometry analysis of major histocompatibility complex (MHC) molecules, epithelial markers, and adhesion receptors in CLECs. One representative is shown out of five CLEC cell lines tested. ∗Cells were permeabilized for staining of intracellular antigens. (B) Schematic of identified isoforms of human leukocyte antigen-G (HLA-G), adapted from Le Discorde et al. (21). There are four membrane bound isoforms and three soluble ones, HLA-G1 and G5 being the full-length isoforms, respectively. Information on alternative splicing of HLA-E is not yet available. (C) Isoforms 1, 5, and 7 of HLA-G mRNA were consistently detected in all five CLEC lines; other isoforms were detected at varying levels across CLEC cell lines from different donors. HLA-E mRNA was also detected consistently. (D) Western blot analysis of HLA-G/E protein expression in CLECs. For HLA-G, a product of approximately 36 kDa (predicted to be isoform 5) was clearly evident in the cell lysate along with a secreted product of approximately 17 kDa (predicted to be isoform 7) in culture medium. For HLA-E, a product of approximately 42 kDa was detected in cell lysates. No secreted HLA-E was detected. JEG3, choriocarcinoma line as a control.
Further analysis of the HLA-G expression was performed with five primary CLEC cell lines from different donors. HLA-G is predicted to have seven protein isoforms based on alternative splicing. Isoforms 1–4 are predicted to be membrane bound and isoforms 5–7 are soluble/secreted (Fig. 2B) (28,31). The mRNA splice variants of HLA-G expressed by CLECs were identified using primer sets targeting specific exons/introns of the HLA-G gene by RT-PCR (Table 1). All of the CLEC lines expressed mRNA for HLA-G isoforms 1, 5, and 7. One cell line expressed isoform 3, four expressed isoform 2/4, and three expressed isoform 6. HLA-E mRNA was also detected consistently in the five CLEC lines (Fig. 2C).
HLA-G and HLA-E protein expression was analyzed in both the cell lysates and culture supernatants of CLECs. The choriocarcinoma cell line JEG3, known to express high levels of HLA-G and E, was used as a positive control (6) (Fig. 2D). For HLA-G, a product of approximately 36 kDa (predicted to be isoform 5) was clearly evident in the cell lysate along with a secreted product of approximately 17 kDa (predicted to be isoform 7) in culture medium. For HLA-E, a product of approximately 42 kDa was detected in cell lysates. No secreted isoforms of HLA-E were detected.
The fact that HLA-E was found in all cells that expressed isoforms of HLA-G is consistent with reports that HLA-E is complexed with the HLA-G signal sequence-derived nonamer (17).
CLECs Inhibit Allogeneic Immune Responses In Vitro
The relative immunoregulatory activity of selected HLA-G/E expressing CLECs was tested using T-lymphocyte proliferation assays. These involved an in vitro surrogate for allogeneic immune responses that employs mixed dendritic cells and T lymphocytes from two genetically unrelated donors to stimulate a mixed leukocyte reaction (MLR) (15). MLR combined with mitogen stimulation assays was employed to test the immunoregulatory activity of culture medium conditioned by CLECs. Conditioned media (supernatants) from CLEC cell lines completely suppressed T-cell proliferation induced by MLR or potent T-cell mitogens (Fig. 3Ai and ii, respectively).

Cord lining epithelial cells inhibit alloreactive T-lymphocyte proliferation. (A) i) Culture supernatants from four CLEC cell lines significantly impaired responses of T lymphocytes in an allogeneic mixed leukocyte reaction (MLR). ii) CLEC supernatants block T-cell responses upon stimulation with the mitogen phytohemagglutinin (PHA). p-Values shown have been corrected (Bonferroni) for multiple comparison. (B) i) Schematic of transwell culture system for comparing membrane bound versus secreted regulatory factors. ii) Coculturing CLEC with MLRs resulted in a significant inhibition of allogeneic T-cell proliferation, through both direct or transwell coculture (cells in transwells are indicated with brackets). Proliferation was evaluated as the ratio of proliferating T cells (CFSElowCD4+) to the entire CD4+ population (data was from six independent MLR experiments). ∗∗p < 0.01 by paired Student's t-test, after Bonferroni correction. CFSE, carboxyfluorescein succinimidyl ester. (C) With fixed cell numbers of T cells and DCs in the MLRs, titrated cell number of cocultured CLECs resulted in a dose-dependent response in inhibition of T-cell proliferation. (D) Immune suppressive effects of CLEC supernatant was reversed by anti-HLA-G blocking antibody but not by an isotype control antibody. Counts per minute (cpm) readings were done with triplicate wells. Error bars represent mean ± SEM.
Transwell coculture experiments were designed to test if the immunoregulatory activity of CLECs was due to secreted products (Fig. 3Bi). In six independent experiments employing different human donors, CLECs significantly reduced the levels of MLR induced proliferation via secreted products and in direct coculture (p < 0.01 by paired t-test with Bonferroni correction) (Fig. 3Bii). When CLECs were separated from the MLR by a 200-kDa cut-off permeable membrane, the proliferation of alloreactive T cells remained significantly impaired. This and the direct coculture results demonstrated that the immunoregulatory activity was mediated by secreted factor(s) from CLECs. Titration of the CLEC cell number against fixed numbers of DCs and CD4+ T cells indicated that the immunoregulation observed was dose dependent. The CLECs, with as few as 5,000 CLEC cells per well in 1-ml cultures, significantly inhibited 1 × 106 alloreactive CD4+ T lymphocytes (Fig. 3C).
To test if soluble HLA-G contributed significantly to the immunoregulatory activity described, a functional assay using anti-HLA-G antibody was conducted with allogeneic MLR. The anti-HLA-G antibody significantly reduced the inhibitory effects of CLEC-conditioned culture supernatants, resulting in T-cell proliferation almost approaching control levels (Fig. 3D). Anti-HLA-G antibody alone (in DMEM control medium) induced a small increase in T-cell proliferation in the MLR. These data strongly support the link between the CLEC immunoregulatory function and the expression of secreted isoforms of HLA-G.
We detected no significant increase in the numbers of CD4+ cells staining positively for CD4/CD25/FoxP3 in the cultures containing CLECs, where MLR responses were clearly impaired (Fig. 4A).
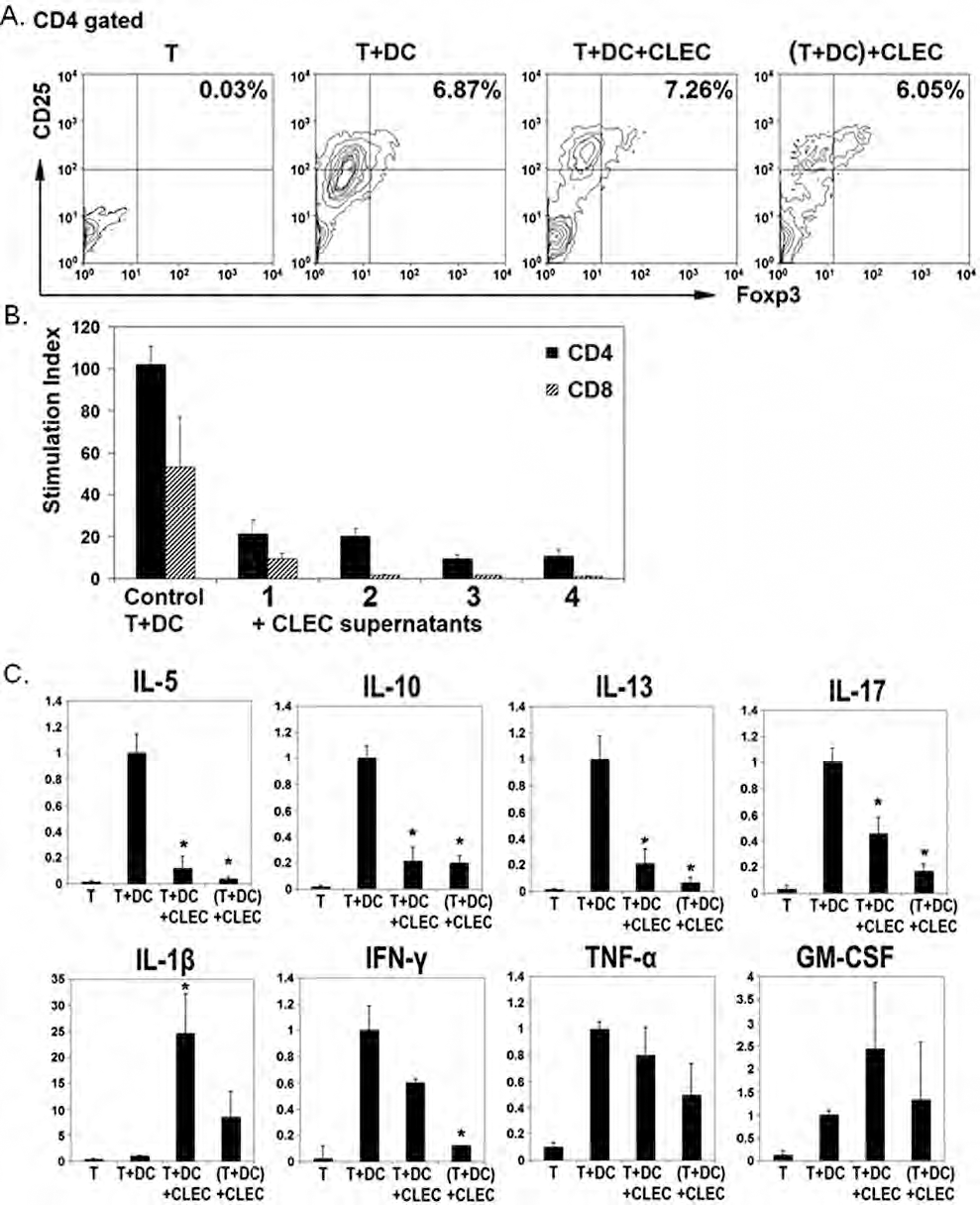
Cord lining epithelial cells modulate the cytokine environment in mixed leukocyte reactions. (A) The upper right-hand quadrant shows the population of CD4+CD25highFoxp3+ regulatory T cells. CLECs did not induce expansion of this regulatory T-cell population in the cocultured MLR. One representative is shown from three independent experiments. Foxp3, foxhead box p3. (B) CLEC-conditioned culture supernatants inhibited proliferation of alloreactive CD4+ and CD8+ T cells. CD4+ T cells (i) responded with higher level of proliferation than CD8+ T cells (ii). Stimulation index shows ratio of [3H]thymidine uptake of stimulated over unstimulated T cells (mean ± SEM). (C) Cytokine concentrations at day 7 of MLR were measured by multiplex bead suspension array. Values of each experimental condition were normalized to the cytokine concentrations in the MLR control (T+DC). ∗p < 0.05 by Wilcoxon signed-rank test, after Bonferroni correction. Data shown were from three to five independent experiments, each with duplicate cultures. IL, interleukin; IFN, interferon; TNF, tumor necrosis factor; GM-CSF, granulocyte-macrophage colony-stimulating factor.
The CLEC conditioned culture supernatants inhibited the proliferation of both CD4+ and CD8+ T cells in MLRs, with consistently lower CD8+ allo-induced proliferation observed (Fig. 4B). Statistically significant inhibition was present only for CD4+ cells. This is consistent with previous reports indicating that the CD4+ rather than the CD8+ T-lymphocyte is the principal responder to allogeneic stimuli in vitro (30), although a role for CD8+ cells may still be present.
Bioplex cytokine arrays were utilized to measure and compare the expression of pro- and anti-inflammatory cytokines in MLRs containing CLECs versus controls. CLECs cocultured with the MLR caused significant decreases in the IL-5, IL-10, IL-13, and IL-17 concentrations in both direct and transwell cocultures. CLECs also caused a decrease in interferon-γ (IFN-γ), which was statistically significant in transwell but not direct cocultures. For IL-1β, CLECs caused an increase, which was significant in direct but not transwell cocultures. GM-CSF and tumor necrosis factor-α (TNF-α) levels in the MLR did not change significantly upon the addition of CLECs (Fig. 4C). In summary, CLECs seemed to down-regulate several proinflammatory cytokines of both the Th1 and Th2 pathways in the MLR, except for some increase in IL-1β.
The data on IL-10, together with the flow cytometry profiles, suggest that CLECs did not work through the induction of CD4+CD25highFoxp3+ regulatory T lymphocytes.
CLECs Survive for Extended Periods as Xenotransplants in Immunocompetent Mice
Our in vitro observations indicate that CLECs have the potential to regulate and dampen human T-lymphocyte responses engendered as part of an allogeneic immune response. In vivo studies based on the xenotransplantation of these cells into immunocompetent Balb/c mice have further indicated their remarkable immunoregulatory potential. Human keratinocytes were used as a control human primary epithelial cell type, which share an epithelial phenotype with the CLECs (36) but do not express the HLA-G and HLA-E molecules. CLECs and keratinocytes were stably transduced with lentiviral constructs and expressed trackable fluorescent transgenes of DsRed2 and eGFP, respectively.
Significant numbers of DsRed2-tagged CLECs could be visualized on the transplanted membranes 14 days later while the eGFP signal had completely disappeared from the keratinocyte grafts (Fig. 5A). Earlier time points revealed rapid clearance of keratinocytes from the transplanted graft. Within 12 h, we observed a significant reduction in the numbers of eGFP-tagged keratinocytes on the transplanted membrane. By 48 h, we detected very few eGFP-positive cells (Fig. 5Bi). Moreover, with mixed CLEC/keratinocyte-coated membranes, large numbers of eGFP-tagged keratinocytes were observed 48 h post-xenotransplantation, suggesting that factors produced by CLECs afford nonspecific protection to other proximal human cell types (Fig. 5Bii).

Cord lining epithelial cells survive in immunologically competent mice and protect proximal human cells from acute xenorejection. (A) Lentiviral transfected CLECs and keratinocytes, expressing DsRed2 and eGFP, respectively, were visualized using fluorescent microscopy (Olympus IX70), before transplantation (in culture), and from excised grafts after 2 weeks. Red fluorescence indicates the presence of CLECs up to 14 days posttransplantation, whereas the green fluorescence of normal human keratinocytes was not detected by scanning the entire graft. (B) i) Examination of grafts at earlier time points showed that keratinocytes dismissed rapidly during the early stages of xenotransplantation and were absent by 48 h, while CLECs persisted through all time points tested (N = 4). ii) Cotransplantation with CLECs improved survival of keratinocytes in the xenotransplantation mouse model. A greater number of eGFP-positive keratinocytes could be found in the mixed cell graft than in the graft with keratinocytes alone (i), up to 2 days posttransplantation. eGFP-positive keratinocytes localized in close proximity with DsRed2-positive CLECs in the graft (N = 2). (C) Reestablished CLEC cultures from the excised graft demonstrated the continuing viability of the transplanted cells. Differential interference contrast (DIC) images indicated the presence of nonfluorescent cells in these cultures, which might be either dead or infiltrating murine cells.
The viability of the grafted CLECs was tested by the reestablishment of in vitro cultures. In vitro cultures were reestablished from grafted CLECs taken from Balb/c mice 12 and 48 h and 14 days posttransplantation. These data would rule out the possibility of false-positive results generated by fluorescence residues from dead cells or by neighboring murine cells that have internalized CLEC cellular debris (Fig. 5C).
CLECs Expressing Human Insulin Regulate STZ-Induced Hyperglycemia In Vivo
To complement our studies employing fluorescent transgenes, we used an in vivo disease model to test the utility of CLECs in the delivery and expression of a candidate therapeutic transgene. Human insulin has previously been shown to regulate STZ-mediated hyperglycemia in rodent and porcine models (7,11,32). We employed CLECs stably expressing human insulin (CLEC-INS for short) in the treatment of STZ-treated SCID mice, which were utilized based on a stable increase in serum glucose levels to higher than 14 mM for 3 days. CLECs were transfected with a lentiviral vector (32) to coexpress two genes driven by a constitutive human immunodeficiency-based lentivirus and murine stem cell retrovirus (HIV/MSCV) promoter: the human proinsulin gene and eGFP gene linked by an internal ribosome entry site (IRES), which served as a marker for successfully transfected/engrafted cells. The transgene human proinsulin was modified to allow cleavage by the ubiquitously expressed protease, furin (13). This gene transfer technique resulted in the efficient and stable expression of human insulin and eGFP in CLECs. After 10 culture passages posttransfection, CLECs remain strongly positive for eGFP (Fig. 6Ai). Moreover, human C-peptide was detected (Fig. 6Aii), indicating that these cells secrete mature, processed human insulin.
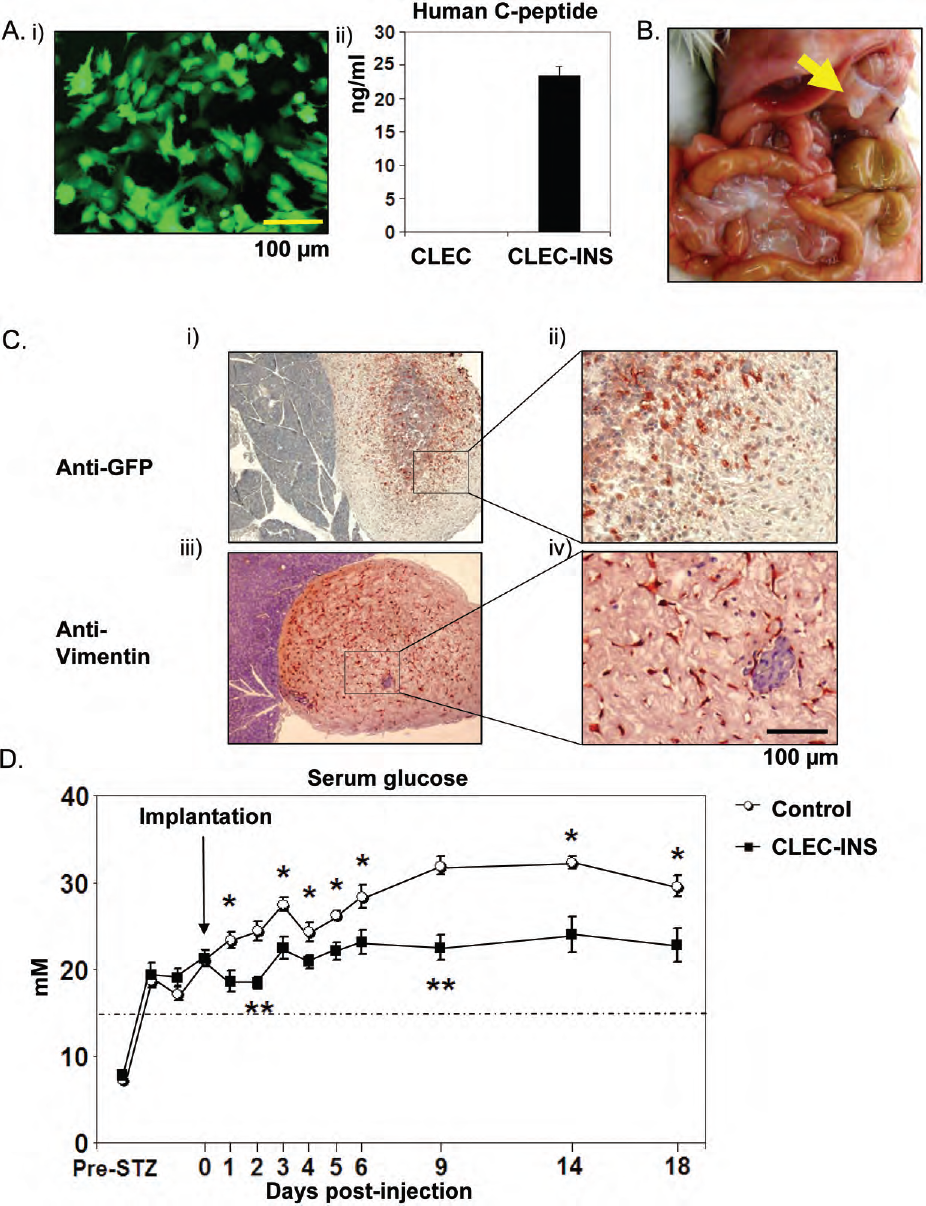
Cord lining epithelial cells mediate delivery of human proinsulin in streptozotocin (STZ)-treated mice. (A) i) Fluorescent microscopic image of human insulin transfected CLEC cells (CLEC-INS) in culture, showing positive eGFP expression. ii) Human C-peptide was detected in CLEC-INS culture supernatants (48-h incubation) at above 20 ng/ml, but not in control CLECs, indicating furin-based processing of expressed proinsulin (N = 3). (B) Necropsy of STZ-treated hyperglycemic severe combined immunodeficient (SCID) mice. An opaque cluster (arrow) was observed on the pancreas in mice injected with CLECs (7 out of 8 mice). (C) Histological analysis of pancreas from CLEC-INS-treated mice. A red/brownish color indicates positive staining for GFP (i and ii) or against human vimentin (iii and iv). (D) Serum glucose levels of the STZ-induced diabetic mice treated with CLEC-INS or control medium (mean ± SEM). Day 0 indicates the time point when CLECs or control medium were injected. N = 5 for medium control, and N = 8 for CLEC-INS. ∗p ≤ 0.05, ∗∗p ≤ 0.01 by Student's t-test. Dashed line marks 14 mM, which was used as criterion for hyperglycemia.
Ten million insulin-expressing CLECs were transplanted into mice pretreated with the β-cell lytic agent STZ to induce pancreatic damage and hyperglycemia (26). We opted for an intraperitoneal delivery of the CLECs based on the anatomical location of the pancreas. A medium-only control group was included. Necropsy of the mice 3 weeks posttransplantation revealed an opaque cluster of cells around the pancreas of the CLEC-INS recipients (7 out of 8 mice) that was absent in the medium-treated controls. A representative CLEC-INS mouse is shown (Fig. 6B). Further analysis of these structures by immunohistochemistry using anti-GFP (Fig. 6Ci, ii) or anti-human vimentin (Fig. 6C iii, iv) antibodies indicated that these opaque clusters were composed of transplanted CLECs.
The statistical significance of differences in the hyperglycemia levels between treatment and control groups was tested using a general linear model for repeated measures. We observed a statistically significant decrease in the serum glucose level for mice that received CLEC-INS from a peak of 32 mM in controls to a stabilized level of approximately 22 mM over 18 days (p = 0.002) (Fig. 6D).
Discussion
In the present study, we have described the immunological characterization of a novel human umbilical cord lining-derived cell. We then sought to address a key issue in cell transplantation; namely, the immune-mediated rejection of cells from genetically unrelated donors.
We have characterized the expression of HLA-G in CLECs, and shown that CLECs secreted soluble HLA-G and suppressed mitogenic and allogeneic T-cell responses. HLA-G expression has been reported in MSCs derived from bone marrow (40,41), Wharton's jelly (20), and amniotic membrane (45). However, these studies either focused on the two full-length isoforms or only distinguished between membrane and soluble forms of HLA-G. There have also been some indications that human amniotic epithelial cells (hAECs) may also express HLA-G (14,25). However, HLA-G expression in isolated hAEC cell lines has not been characterized in vitro. The present study has presented novel data on the analysis of all seven known isoforms of HLA-G in CLECs.
In addition, the present study has also demonstrated clearly, using a functional assay, that the secreted soluble HLA-G played a key role in the immunoregulatory properties of CLECs. Our study also supports the suggestion that, in contrast to bone marrow-derived MSCs, this immunoregulation is less likely to be mediated by an induction of CD4+CD25highFoxP3+ regulatory T cells (41).
The immunological properties of CLECs characterized in this study are consistent with reports on other types of extra-embryonic tissue derived cells, such as Wharton's jelly stromal cells (46), umbilical cord perivascular cells (10), amniotic membrane mesenchymal (24) and epithelial cells (23), and amniotic fluid stem cells (42). Many of these cells, and CLECs, consistently express MHC-I but not MHC-II antigens, with the exception of some umbilical cord perivascular cells which were reported to be MHC-I negative (39). CLECs also share with the above mentioned cells the ability to inhibit lymphocyte proliferation in vitro. These immunological properties might be a common feature associated with their origin from the maternal/fetal interface.
In the present study, the immunomodulatory activity of CLECs was further demonstrated by their survival as xenotransplants in immunocompetent Balb/c mice without any immunosuppression. CLECs survived up to 14 days under the condition of normal xenorejection while normal human keratinocytes were cleared within 48 h. In addition to the detection of fluorescent markers, the CLECs could be retrieved from the xenograft and recultured, and maintained their morphology and transgene marker expression. Most previous studies on in vivo survival of cell transplantations have used the persistence of biochemical markers to indicate survival of the transplanted cells. These have included detection of transplanted cell-specific proteins/DNAs (25) or prelabeled molecular markers (38), to demonstrate survivability. One of the limitations of these findings has been the possibility of the persistence of these markers in dead (but not degraded) cells or the possible internalization of cellular debris by adjacent host cells. The current study has provided stronger evidence by retrieving these cells from the mice, and then establishing viable cell cultures from these retrieved cells.
Cotransplanted CLECs also provided protection to the human keratinocytes and prolonged their survival significantly. This suggests that CLECs may provide a protective environment to favor survival, as has been reported with MSCs as well (3,44), and thus may be of potential use in this manner.
Finally, in a pilot study, we transfected CLECs with a human proinsulin gene to test a potential therapeutic application of these cells. We found that CLECs that constitutively secrete human insulin effected a significant reduction in serum glucose levels in STZ-treated hyperglycemic mice over 18 days. The serum glucose levels in CLEC-INS-treated mice were not reduced to the minimal range of approximately 5 mM. This may be explained by the fact that human insulin was used to treat STZ-induced hyperglycemia in mice (29).
Following the intraperitoneal injection of CLECs into SCID mice with STZ-induced hyperglycemia, we observed a natural clustering of the CLECs proximal to the pancreas. This was an unexpected observation that may indicate the possibility of specific chemotaxis of CLECs to the STZ-damaged tissue (i.e., the pancreas) as part of a tissue repair/regenerative process.
In conclusion, the combination of the in vitro and in vivo data presented, plus the ease of CLEC derivation, demonstrates the potential of CLECs as a new candidate for cell transplantation.
Footnotes
Acknowledgments
We would like to acknowledge Kae Siang Ngo for his assistance with the xenograft experiments, Desmond Chan for his assistance with the thymidine proliferation assays, Hwan Chour Han for his assistance with immunohistochemistry, Ooi Shu Qin for her assistance with statistics, Professor Didier Trono (University of Lausanne, Switzerland) for provision of the construct used in our in vivo survival experiments for labeling of the transplanted cells, and Professor Ann Simpson (University of Technology, Sydney) for provision of the construct used in our in vivo gene therapy experiments on streptozotocin (STZ)-treated mice. This study was supported by research grants from the SingHealth Foundation (SHF/FG347P/2006), National Medical Research Council (NMRC R-176-000-069-213, NMRC/TCR/005-NUS/2008), National University of Singapore Department of Surgery (C-176-000-003-001), and Kidney Dialysis Foundation, Singapore (R-176-000-116-720). Professor Toan Thang Phan is a founder and shareholder of CellResearch Corporation Pte, Ltd.