
Abstract
There have been various forms of mesenchymal stem cell-like (MSC-like) cells isolated from umbilical cords (UCs). The isolation of umbilical cord lining stem cells (ULSCs) may be of great value for those interested in a possible treatment to several disease/disorders. Unlike umbilical cord blood cells, these cells are unique because they can be expanded to therapeutically relevant numbers and cryopreserved for several different uses. Here we efficiently isolate stem cells from a small segment of pre- and postnatal UCs, and obtain therapeutically relevant amounts of ULSCs within 3 weeks. We demonstrate their growth potential and characterize them using immunocytochemistry, flow cytometry, and RT-PCR. In addition, we differentiate ULSCs into multiple lineages. Pre- and postnatal ULSCs are morphologically similar to mesenchymal stem cells (MSCs) and easily expand to greater than 70 population doublings. They express pluripotent markers Oct4 and nanog at the protein and RNA level. Flow cytometry demonstrates that they express markers indicative of MSCs in addition to high SSEA-4 expression. ULSCs are easily differentiated into osteogenic, adipogenic, chondrogenic, cardiogenic, and neurogenic cells. Pre- and postnatal ULSCs are characteristically similar in respect to their growth, marker expression, and plasticity, demonstrating they are highly conserved throughout development. ULSCs have phenotypic and genotypic properties of MSCs. These studies demonstrate the therapeutic potential of an otherwise discarded tissue. They are a perfect HLA match for the donor and an excellent match for immediate family members; therefore, they may serve as a therapeutic cell source.
Keywords
Introduction
Stem cells have been isolated from all developmental periods of life and are thought to participate in tissue regeneration/repair. Hematopoietic stem cells (HSCs) isolated from bone marrow has been used for years in order to reconstitute the bone marrow of recipients suffering from hematological disorders (2). Since its initial clinical use 20 years ago, umbilical cord blood (UCB) has been considered an alternative source of HSCs (12). In addition, it is considered a better source of HSCs due to its ease of collection, more tolerance of HLA mismatches, less graft-versus-host disease, and may be used allogeneically (16,24,25). Most importantly, the umbilical cord and its blood are usually considered otherwise discarded tissue, avoiding any ethical issues. However, due to the low number of HSCs obtained, it is only clinically applicable for infants/children with hematological disorders. In theory, a fewer number of HSCs require a greater length of time to engraft and the patient might die as a result of secondary factors such as infections due to the lack of a functional hematopoietic system (19).
Besides HSCs, one of the most widely studied adult stem cells for regenerative medicine are mesenchymal stem cells (MSCs). In the adult, MSCs are found at higher density in the marrow cavity but the number decreases with age, making it difficult to create substantial amounts of therapeutic relevance (33). MSC-like cells can be isolated from various sources such as the umbilical cord (1), placenta (23), and amniotic fluid (8). It is well documented that MSCs are immunosuppressive (3,15,17), making them excellent candidates for treatment of many disease states. For instance, transplantation of fully mismatched allogeneic fetal liver-derived MSCs into an immunocompetent human fetus with osteogenesis imperfecta was tolerated and engrafted into bone (18).
MSCs are presently the forerunner for cell replacement strategies as a result of extensive research demonstrating their capability of self-renewal, differentiation, engraftment, and ability to ameliorate symptoms in animal models of numerous diseases (6,29,30,34,35). Thus, numerous laboratories are performing clinical safety and efficacy studies using MSCs for the treatment of a number of pathological conditions such as heart failure (22), spinal cord injury (20), and bone and cartilage repair (31).
There have been various forms of MSC-like cells isolated from the umbilical cord termed umbilical cord matrix stem cells (UCMSCs), umbilical cord perivascular cells (UCPVs), umbilical cord stroma cells (UCSCs), and most recently cord lining membrane-MSCs (CLMSCs) (13,14,27,32). The cells isolated in this report we term umbilical cord lining stem cells (ULSCs), which may be of great value for individuals who are interested in a possible treatment to several disease/disorders. Unlike umbilical cord blood cells, these cells are unique as they can be expanded in culture to therapeutically relevant numbers and cryopreserved for several different uses. The collected cells are a perfect HLA match for the donor and an excellent match for immediate family members (26).
Here we describe a rapid and efficient method to isolate stem cells from the umbilical cord lining (ULSCs) using the explant method. We isolate greater than 4 × 106 cells within 10 days using only 6—8 cm of umbilical cord, which can be further expanded within 2 more weeks to therapeutically relevant numbers (2.0 × 108). We characterize ULSCs isolated from pre- and postnatal umbilical cord. Both pre- and postnatal ULSCs express pluripotent markers Oct4 and nanog and have similar characteristics to MSCs. In addition, pre- and postnatal ULSCs are easily differentiated into osteo, chondro, adipo, neural, and cardiac cell types. These are the first known studies characterizing stem cells isolated from pre- and postnatal umbilical cord, which may have future implications for stem cell banking. Interestingly, these data demonstrate that the umbilical cord is highly conserved throughout development.
Materials and Methods
Ethics and Consent
Umbilical cords were obtained from DV Biologics LLC (Costa Mesa, CA). All umbilical cords were obtained from various donors after informed consent as approved by an institutional regulatory board (IRB) (DV Biologics). Prenatal umbilical cords were obtained from donors of elective or spontaneous abortions. Postnatal umbilical cords were obtained from donors of full-term pregnancies.
Umbilical Cord Processing
All procedures were performed aseptically in a class II biosafety cabinet (Class 100). Prior to commencing, six six-well dishes (Nunc, Rochester, NY) were precoated with human fibronectin (1 μg/ml) (Sigma Aldrich, St. Louis, MO). After obtaining 6-8 cm of umbilical cord (UC) according to approved consented method, the UC was washed with Hanks basic salt solution (HBBS) (Hyclone, Rochester, NY) followed by a wash in a 10% betadine solution with HBSS for 1 min. UC were cut into 0.5—1-cm cross sections, which were then cut longitudinally, avoiding the vein and arteries. The vein and arteries were dissected out along with any excess gelatinous material. The resulting dissected UC lining was placed into fresh HBSS for explanting.
Umbilical Cord Lining Explants
One dissected cord lining piece was cut into 3—4 strips with the gelatinous side up. Two to three pieces of UC were seeded onto human fibronectin (Sigma Aldrich)-coated six-well dishes. A 22 × 22-mm sterile coverslip (Fisher Scientific; Pittsburg, PA) was placed on top of the cord lining and 1.5 ml of media was added into the well. Explants and cells were grown in DMEM (Gibco, Carlsbad, CA), 15% fetal bovine serum (FBS) (Hyclone), penicillin/streptomycin, Glutamax, MEMNEAA, and MEM vitamins (all from Gibco). All six-well dishes were kept in 5% CO2 incubators at 37°C immediately after completing explants.
Cell Culture
Media change was performed two to three times a week. At 70—80% confluency cells were detached using enzymatic digestion (Gibco) and plated onto human fibronectin-coated T225 flasks at a density of 1,000 cells/ cm2.
Colony-Forming Units (CFU) Assay
For colonies, cells were plated at a density of 100 cells/10-cm dish in the above described media. After 14 days, cells were fixed and stained in a 9% crystal violet methanol solution for 1 min. Cloning efficiency was estimated as percentage of cells that generated clones from total cell number/dish.
Growth Kinetics
For population doublings (PD), cells were cultured on 25-cm2 flasks, harvested, counted, and replated when they reached 70—80% confluency. Cell culture was terminated when cell population failed to double after 1 week of culture. Population doubling was calculated using the formula PD = [log10(N 1) — log10(N 0)/log10(2)], where N1 is cell number at harvesting and N 0 is cell number at plating, as previously described (7).
Immunocytochemistry (ICC)
Cells were fixed in 4% paraformaldehyde (PFA) and stored at 4°C. After permeabilization in 0.1% of Triton X-100 (Promega, Madison, WI) for 5 min and blocking in 10% donkey serum (Jackson Immunoresearch, West Grove, PA)/0.5% Triton X-100 for 1 h, primary antibody diluted in 1% donkey serum was applied overnight at 4°C. Cells were incubated with secondary antibody in blocking buffer for 1 h at room temperature (RT). Cells were counterstained with DAPI (Molecular Probes, Carlsbad, CA) and mounted with Fluoromount-G (Southern Biotech, Birmingham, AL). Primary antibodies used were: Oct3/4 clone H-134 (Santa Cruz Biotech, Santa Cruz, CA), nanog (ReproCell, Tokyo, Japan), SSEA-4 (Millipore, Billerica, MA), CD90 (Thy-1) (Millipore), cardiac sarcomeric actin (Abcam, Cambridge, MA), myosin heavy chain (Upstate, Billerica, MA), desmin (Millipore), troponin I (Abcam), connexin 40 (Santa Cruz Biotech), nestin (millipore), A2B5 (Millipore), O4 (Millipore), β-tubulin III (Sigma Aldrich). Secondary antibodies Alexa 488 and Alexa 594 (Molecular Probes) were used. For negative controls incubation without primary antibody and with corresponding specific nonimmune immunoglobulins (Santa Cruz Biotech) was used. Staining was analyzed using an Olympus IX81 inverted microscope and SlideBook software.
Flow Cytometry
Cells were detached, filtered through a 40-μm strainer, pelleted, resuspended in MEM + HEPES (Gibco) with 2% BSA, and quantified. Directly conjugated antibodies used were: CD105, CD106, CD166, CD117, CD90, CD44, CD45, CD34, CD19, HLA-ABC, HLA-DP DQ DR (Serotec, Oxford, UK), CD133 (Miltenyi Biotech, Germany), LIN, and CD73 (BD Pharmingen, San Diego, CA). For anti-SSEA-4 and anti-STRO-1 staining (Millipore) secondary antibody goat anti-mouse IgG + IgM-APC (Jackson Immunoresearch) was used. After staining, cells were fixed with 4% paraformaldehyde and analyzed using CyAn ADP Analyzer 9 color (Beckman Coulter, Fullerton, CA). Histograms were generated by using Flowjo software (Treestar Inc, Ashland, OR).
PCR Analysis
To analyze gene expression profile, cells were collected in RLT buffer (Qiagen, Hilden, Germany) and stored at −80°C. NT2 cells used as a control for pluripotency genes were purchased from ATCC (Manassas, VA). Total RNA from prenatal brain and cardiac tissue was purchased from DV Biologics. Total RNA was isolated with the RNeasy Plus kit (Qiagen). RNA (300 ng) was reverse transcribed using LongRange Reverse Transcriptase from Qiagen. PCR was performed applying GoTaq® Hot Start polymerase (Promega, Madison, WI) in a 25-μl total volume and C1000™ Thermal Cycler (Bio-Rad, Hercules, CA). Table 1 shows gene-specific primers that were used in end point PCR, utilizing 1 μl of cDNA product. “No RT” controls were included in each experiment and showed no contamination with genomic DNA. Agarose gels were scanned using Bio-Rad Molecular Imager ChemiDoc XRS+ System. For quantification purposes, the bands were analyzed applying Quantity One 1-D Analysis Software. The band intensity of all examined genes was normalized to β-actin level.
RT-PCR Primers Used in This Study

Cell Differentiation
Adipogenic Differentiation. Cells at passages 1—4 were plated at a density of 4,000cells/well in 12-well plates in regular culture medium. At 90—100% confluency, following components were added to the media for adipogenic induction: 0.5 μM isobutylmethylxanthine, 50 μM indomethacin, and 0.5 μM dexamethasone (all from Sigma Aldrich). Media was changed every 2—3 days for 21 days. Control cells were kept in regular culture medium. At 21 days cells were fixed with 4% PFA and stored at 4°C in phosphate buffer saline (PBS) until staining. Staining was performed using 0.3% Oil Red O solution (Sigma Aldrich).
Osteogenic Differentiation. Cells plated on 12-well dishes were switched to osteogenic differentiation medium (HyClone) according to the manufacturer's protocol when 90—100% confluent. After 21 days of induction cells were fixed in 4% PFA and stained with 2% Alizarin Red S (Sigma).
Chondrogenic Differentiation. Cells were placed in either control media or chondrogenic differentiation medium according to the manufacturer's protocol (Hyclone). Briefly, 300,000 cells/15-ml tube were pelleted and control or chondrogenic differentiation medium was added. After 28 days, pellets were fixed with 4% PFA. Pellets were sunk in 25% sucrose solution and frozen embedded 48 h after in OCT compound (Sakura Finetek, Torrance, CA). Pellets were sectioned at 10 μm and stained with 1% Alcian Blue (Sigma Aldrich) and counterstained with nuclear fast red (Sigma Aldrich) using standard protocols.
Cardiac Differentiation. Cells were plated on 12-well dishes at a density of 4,000 cells/well in control media until 80—90% confluent. For cardiac differentiation cells were treated with 5-azacytidine at a final concentration of 10 μM (Sigma Aldrich) for a 24-h period and then at 7 days for another 24-h period. After 14 days cells were fixed with 4% PFA and stored at 4°C in PBS until staining.
Neural Differentiation. Cells were plated on Matrigel-coated (BD Biosciences, San Diego, CA) 12-well dishes at a density of 4,000 cells/well. For differentiation, cells were placed into a 50:50 blend of control media and neural induction media, which consisted of DMEM/F12, 10% serum replacement, N2 supplement, glutamax, MEM NEAA, MEM vitamins (all from Gibco), ITS premix (BD Biosciences), and growth factors SHH (100 ng/ml) (R&D Systems, Minneapolis, MN), FGF8 (100 ng/ml), and bFGF (20 ng/ml) or EGF (20 ng/ml), bFGF (20 ng/ml), and PDGF (20 ng/ml) (all from Invitrogen). The following day cells were placed into 100% neural induction media. On the seventh day, 3 μM retinoic acid (RA) (Sigma Aldrich) was added for 3 consecutive days. Neural induction conditions were then changed to GDNF (20 ng/ml) (Biosource), BDNF (20 ng/ml) (Biosource), and 200 μM ascorbic acid addition for the cells on SHH, FGF8, and bFGF, and EGF alone for cells on EGF, bFGF, and PDGF for an additional 14 days. Cells were fixed with 4% PFA and stored at 4°C in PBS until staining.
Karyotyping
Karyotype analyses were performed by Cell Line Genetics using standard cytogenetic protocols. Briefly, G-banding technique was applied to karyograms produced from at least 20 metaphases.
Results
Growth and Characterization of Pre- and Postnatal ULSCs
We sought to characterize cells isolated from pre-and postnatal umbilical cords in an efficient manner because these tissues are considered otherwise discarded and may be of therapeutic relevance. Our previous experiments using enzymatic digestion of umbilical cords yielded too few cells in order to rapidly and efficiently expand ULSCs (unpublished observations, R.G.). In addition, previous studies have also demonstrated that isolation of stem cells from the umbilical cord using enzymatic digestion yields a small starting population and takes over a month to produce therapeutic relevant cell numbers (13,32). Therefore, we sought to find an efficient method to culture therapeutically relevant numbers of ULSCs. When we used the explant method with coverslips, cells commenced to migrate off of the explants and at 10 days postexplants we obtained an average of 4.9 × 106 cells (1.6 × 106 SD; n = 6) from 6—8 cm of UC. ULSCs display typical MSCs morphology (Fig. 1A, B) and are easily expandable to greater than 70 population doublings within 100 days (Fig. 1C, D). Most importantly, ULSCs can be expanded to therapeutically relevant numbers (>2.0 × 108 cells) within 3 weeks. In order to assess clonogenicity, we performed colony forming unit assays at passage 2, which demonstrated that ULSCs have a cloning efficiency of 74.25% (SD 7.65%) and 65.75% (SD 4.64%) for pre- and postnatal ULSCs, respectively (n = 4 per group). Both ULSCs populations exhibit diploid cells without chromosomal aberrations at passage 3 (~26 days in culture) as determined by karyotype analysis (Fig. 1E, F). We observed no differences in growth kinetics for pre- and postnatal ULSCs.
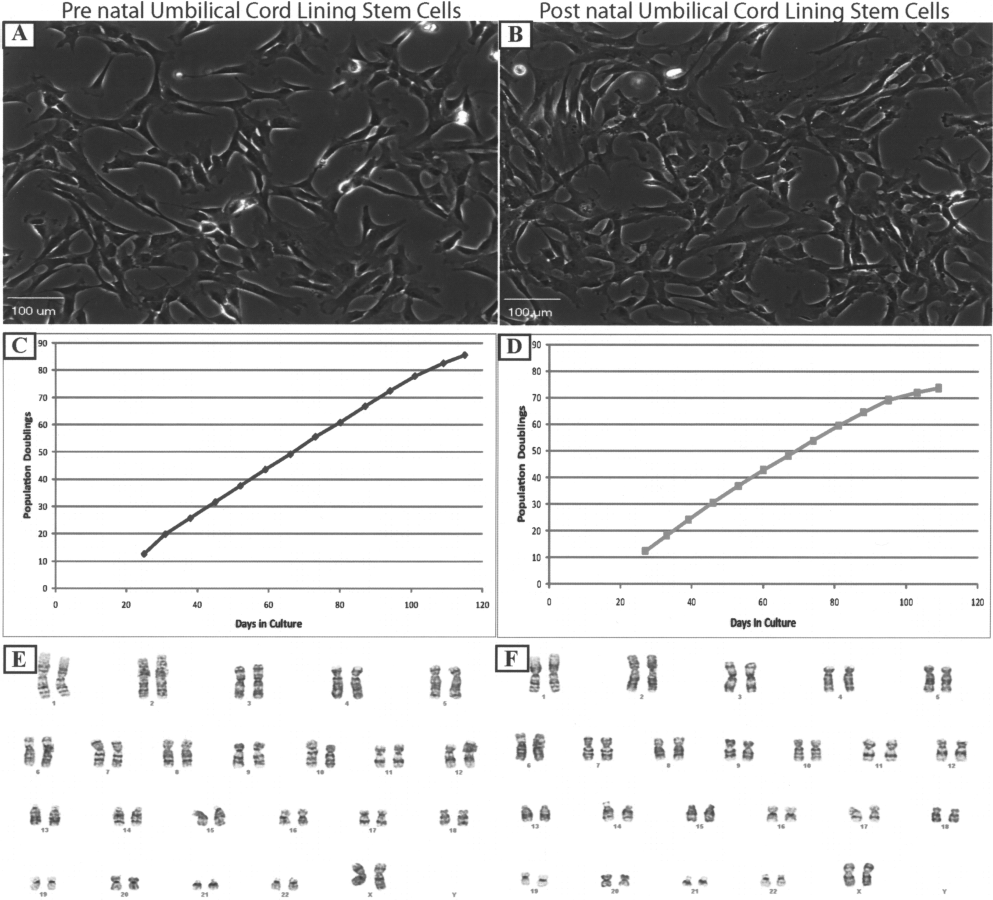
Prenatal (left) and postnatal (right) umbilical cord lining stem cells (ULSCs) have MSC-like morphology, grow rapidly, and exhibit normal karyotype. Phase contrast pictures of prenatal (A) and postnatal (B) ULSCs illustrate MSC-like morphology. Prenatal (C) and postnatal (D) ULSCs growth curves illustrate rapid growth. Karyotype analysis of ULSCs demonstrates no chromosomal aberrations (E, F). (A, B) Original magnification 10x; scale bars: 100 μm.
In order to characterize pre- and postnatal ULSCs we performed RT-PCR, immunocytochemistry (ICC), and flow cytometry. RT-PCR analysis of pre- and postnatal ULSCs demonstrates that these cells express the pluripotent markers Oct4 and nanog at passage 3 minimum. Similar to a previous publication (14), there was no expression of Sox-2 at any passage (Fig. 2A). The ratio of Oct4 and nanog expression in cultured cells was higher by 15% and 20%, respectively, in prenatal UC when compared to postnatal UC (Fig. 2A). ICC analysis confirmed that ULSCs express pluripotent markers nanog and Oct 4 (Fig. 2B, D prenatal, C, E postnatal). Interestingly, the cells highly expressed the marker stage-specific embryonic antigen 4 (SSEA-4), which is normally found in embryonic stem cells (ESCs) (Fig. 2B prenatal, C postnatal, Fig. 3). Our ICC data support and extend our RT-PCR data demonstrating that ULSCs express pluripotent markers and are of primitive origin. ICC also demonstrates expression of the stem cells marker Thy-1 (CD-90) typical of MSCs on pre- and postnatal ULSCs (Fig. 2D, E). The flow cytometry data show that ULSCs express markers characteristic of MSCs, being positive for CD44, CD73, CD90 (Thy-1), CD105, CD106, CD166, STRO-1, SSEA-4, HLA-ABC, and LIN while negative for CD19, CD34, CD45, CD117, CD133, and HLA-DR (Fig. 3). According to our flow cytometry data, there was a negligible amount of CD45-positive cells (<1%) in the postnatal ULSCs. Our flow data supports our ICC and RT-PCR data demonstrating that pre- and postnatal ULSCs are highly conserved throughout development.

Characterization of ULSCs by RT-PCR and immunocytochemistry. ULSCs express pluripotent markers Oct4 and nanog at the RNA level (A). Immunocytochemistry of ULSCs demonstrates expression of pluripotent markers SSEA-4, nanog, and Oct4. In addition, ULSCs express MSCs marker Thy-1 (B and D, prenatal; C and E, postnatal). (A) Lane W, water; lane 1, NT2 cell control; lane 2, prenatal UC tissue; lane 3, postnatal UC tissue; lane 4, prenatal ULSCs passage 1; lane 5, prenatal ULSCs passage 2; lane 6, prenatal ULSCs passage 3; lane 7, postnatal ULSCs passage 1; lane 8, postnatal ULSCs passage 2; lane 9, postnatal ULSCs passage 3. (B—E) DAPI blue. Scale bars: 10 μm.

Flow cytometry of ULSCs. Pre- and postnatal ULSCs express markers characteristic of MSCs. ULSCs are positive for markers CD44, CD73, CD90 (Thy-1), CD105, CD106, CD166, STRO-1, SSEA-4, HLA-ABC, and LIN while being negative for CD19, CD34, CD45, CD117, CD133, and HLA-DR.
Differentiation of Pre- and Postnatal ULSCs
Stem cells are characterized by their plasticity or ability to differentiate into multiple lineages. We therefore undertook studies differentiating ULSCs into multiple cell types. The hallmark of MSCs is their ability to differentiate into adipogenic, osteogenic, and chondrogenic cell types. Using standard protocols, ULSCs induced to differentiate towards adipogenic, osteogenic, and chondrogenic lineages demonstrated histological characteristics of fat droplets found in adipocytes (Fig. 4A), calcium deposits typical of bone (Fig. 4B), and sulfated proteoglycans staining typical of cartilage (Fig. 4C) compared to noninduced control ULSCs. These data clearly demonstrate that ULSCs easily differentiate into mesodermal tissues.

ULSCs easily differentiate into mesoderm cell types. Staining with Oil Red O for fat vacuoles demonstrates differentiation into adipogenic cells when induced compared to noninduced ULSCs (A). Staining with alizarin red S shows calcium deposits typical of osteogenic cell types when induced compared to noninduced ULSCs (B). Staining for sulfated proteoglycans demonstrates differentiation into chondrogenic cell types compared to noninduced ULSCs (C). (A, B) 40x, (C) 10x, inserts left bottom corner 40x. Scale bars: 200 μm.
Pre- and postnatal ULSCs can also be differentiate into cardiac and neural cell types. ICC analysis demonstrates that prenatal (Fig. 5A) and postnatal (Fig. 5B) ULSCs express the cardiac markers desmin, alpha sarcomeric actin, and myosin heavy chain. RT-PCR data support and extend the ICC analysis by demonstrating expression of the cardiac markers troponin T, connexin 43, myosin heavy chain 7, and alpha sarcomeric actin at the RNA level (Fig. 5C). There was no RNA expression of the developmental transcription factor NKX 2.5 in any of the ULSCs tested. Placing pre- and postnatal ULSCs into neural differentiation media causes them to differentiate into neural cell types. Differentiated ULSCs expressed the neural markers nestin, NG2, CNPase, MAP2, and β-tubulin III at the RNA level (Fig. 5D). Nestin, a neural progenitor marker, was only expressed during the beginning of our differentiation experiments and not in control cells (Fig. 5D). ICC analysis demonstrates that prenatal (Fig. 5E) and postnatal (Fig. 5F) ULSCs express the neural cell markers nestin, β-tubulin III, and O4. ICC data support our findings for the ability of ULSCs to differentiate into neural cell types. These data clearly demonstrate that ULSCs are multipotent and can differentiate into multiple cell types.

ULSCs differentiate into cardiac and neural cell types. Immunocytochemistry of prenatal (A) and postnatal (B) ULSCs differentiated into cardiac cells demonstrates expression of cardiac markers desmin, alpha sarcomeric actin, and myosin heavy chain (MHC). RT-PCR demonstrates RNA expression of troponin T, connexin 43, MHC 7, and alpha sarcomeric actin in ULSCs (C). There was no expression of the transcription factor NKX 2.5. RT-PCR data of ULSCs differentiated into neural cell types demonstrates expression of GFAP, NG2, nestin, MAP-2, β-tubulin III, and CNPase (D). Immunocytochemistry of prenatal (E) and postnatal (F) ULSCs differentiated into neural cell types exhibit expression of nestin, β-tubulin III, and O4. (C) Lane 1, undifferentiated postnatal ULSCs; lane 2, cardiac differentiated postnatal ULSCs; lane 3, undifferentiated prenatal ULSCs; lane 4, cardiac differentiated prenatal ULSCs; lane H, heart; lane W, water. (D) Lanes 1—7 postnatal: lane 1, differentiated using SHH/FGF8/bFGF 2 weeks postinduction; lane 2, differentiated using PDGF/EGF/bFGF 2 weeks postinduction; lane 3, differentiated using EGF/ bFGF 2 weeks postinduction; lane 4, undifferentiated control; lane 5, differentiated using SHH/FGF8/bFGF 24 days postinduction; lane 6, differentiated using PDGF/EGF/bFGF 24 days postinduction; lane 7, differentiated using EGF/bFGF 24 days postinduction. Lanes 8—14 prenatal: lane 8, differentiated using SHH/FGF8/bFGF 2 weeks postinduction; lane 9, differentiated using PDGF/EGF/ bFGF 2 weeks postinduction; lane 10, differentiated using EGF/bFGF 2 weeks postinduction; lane 11, undifferentiated control; lane 12, differentiated using SHH/FGF8/bFGF 24 days postinduction; lane 13, differentiated using EGF/bFGF 24 days postinduction; lane 14, differentiated using PDGF/EGF/bFGF 24 days postinduction, lane W, water; lane B, brain. Scale bars: 10 μm.
Discussion
The umbilical cord (UC) serves as a survival system for the developing fetus throughout pregnancy. It is composed of a jelly-like substance that is designed to prevent the compression, torsion, and bending of the three enclosed vessels for bidirectional maternal—fetal blood flow in order to assure appropriate development (4). Changes in the UC during development can cause developmental abnormalities. Here we demonstrate that cells isolated from the UC are highly conserved throughout development. We characterized cells isolated from pre- and postnatal umbilical cord lining (ULSCs) and demonstrate that their growth, marker expression, and differentiation potential are conserved throughout development. In addition, this study documents a rapid and efficient approach to isolating a therapeutically relevant number of stem cells from the umbilical cord lining (ULSCs). These cells have morphological characteristics of MSCs; they form CFUs, display markers indicative of MSCs, and most importantly can be differentiated into multiple cell types. These are the first documented studies demonstrating that cells isolated from the umbilical cord lining are highly conserved during development and may serve for the building of a cell bank for therapeutic purposes, because this is considered otherwise discarded tissue.
Cells isolated from the UCB have been used in transplantation studies for decades. However, their use is limited to infants and small children with hematopoietic malignancies, marrow failure, and immunodeficiency disorders (19,21). Hence, the investigation of other cell sources isolated from the UC. Various groups have isolated cells from UCs which have characteristics of MSCs; however they are heterogenous, which is demonstrated by their varying marker expressions (5,13,14,27,32). Indeed, the differences are due to the different isolation techniques. Regardless, they may all have therapeutic potential. Pre- and postnatal ULSCs isolated using the explant method are rapidly grown to therapeutic amounts (Fig. 1C, D). Other published studies have all used enzymatic digestion in order to obtain cells from the UC except Kita et al. (14).
At the production of greater than 1.0 × 109 cells (passage 3), ULSCs display normal karyotypes (Fig. 1E, F) and express pluripotent markers nanog, Oct4, and SSEA-4 (Figs. 2 and 3) but not Sox-2 when cultured. These findings are similar to a recent publication isolating cells from the subamniotic umbilical cord lining membrane (CLMSCs) (14). However, our ULSCs differ in marker expression. For instance, CLMSCs express CD14, a marker indicative of the innate immune system and of hematopoietic origin. According to the International Society for Cellular Therapy minimal criteria for defining multipotent MSCs, the cells should be CD14 negative (9). CLMSCs also did not express STRO-1, a marker indicative of MSCs that is highly expressed in both pre- and postnatal ULSCs (Fig. 3). Indeed, it has been demonstrated that STRO-1+ cells form only CFUF, have higher rates of adherence, proliferation, and may be a primitive progenitor cell type in the bone marrow (28). Thus, it is clear to state that ULSCs are a different cell population than CLMSCs. ULSCs express high levels of SSEA-4 (99%), a marker of indicative of early embryonic development and stem cells (Fig. 2,3). It has been demonstrated that SSEA-4 can be used to isolate adult MSCs that completely exclude the hematopoietic population (11).
ULSCs display typical MSC characteristics, as demonstrated by flow cytometry, ICC, and their ability to differentiate into bone, fat, and cartilage. Most importantly, pre- and postnatal ULSCs differentiate into multiple cell types (Figs. 4 and 5), which is the hallmark of a stem cell. Several reports have already demonstrated that cells isolated from the UC are multipotent and can differentiate into multiple cell lineages (10,13,14,27,32). However, no reports have demonstrated their ability to differentiate into both neural and cardiac cells within one study, which we have shown in this report. ULSCs displayed markers indicative of cardiac and neural cell types at the RNA and protein level (Fig. 5). These data support the notion that ULSCs are a multipotent stem cell source. Cellular transplantation studies into animal models of various diseases will help elucidate the therapeutic potential and function of ULSCs.
This study documents an efficient approach to isolation and expansion of cells from the umbilical cord lining for potential therapeutic use. These cells are highly conserved throughout their development as demonstrated by growth kinetics, clonogenicity, marker expression, and differentiation potential. Most importantly, these studies document that an otherwise discarded tissue type may be banked and used for therapeutic applications.
Footnotes
Acknowledgments
The authors would like to thank Giorgia Pirino for technical assistance and the Isaias family for their continual support. DaVinci Biosciences has filed a patent application relating to the technology disclosed in this manuscript.