
Abstract
The ability of mesenchymal stem cells (MSCs) to differentiate into neural cells makes them potential replacement therapeutic candidates in neurological diseases. Presently, overexpression of brain-derived neurotrophic factor (BDNF), which is crucial in the regulation of neural progenitor cell differentiation and maturation during development, was sufficient to convert the mesodermal cell fate of human umbilical cord blood-derived MSCs (hUCB-MSCs) into a neuronal fate in culture, in the absence of specialized induction chemicals. BDNF overexpressing hUCB-MSCs (MSCs-BDNF) yielded an increased number of neuron-like cells and, surprisingly, increased the expression of neuronal phenotype markers in a time-dependent manner compared with control hUCB-MSCs. In addition, MSCs-BDNF exhibited a decreased labeling for MSCs-related antigens such as CD44, CD73, and CD90, and decreased potential to differentiate into mesodermal lineages. Phosphorylation of the receptor tyrosine kinase B (TrkB), which is a receptor of BDNF, was increased significantly in MSC-BDNF. BDNF overexpression also increased the phosphorylation of β-catenin and extracellular signal-regulated kinases (ERKs). Inhibition of TrkB availability by treatment with the TrkB-specific inhibitor K252a blocked the BDNF-stimulated phosphorylation of β-catenin and ERKs, indicating the involvement of both the β-catenin and ERKs signals in the BDNF-stimulated and TrkB-mediated neural differentiation of hUCB-MSCs. Reduction of β-catenin availability using small interfering RNA-mediated gene silencing inhibited ERKs phosphorylation. However, β-catenin activation was maintained. In addition, inhibition of β-catenin and ERKs expression levels abrogated the BDNF-stimulated upregulation of neuronal phenotype markers. Furthermore, MSC-BDNF survived and migrated more extensively when grafted into the lateral ventricles of neonatal mouse brain, and differentiated significantly into neurons in the olfactory bulb and periventricular astrocytes. These results indicate that BDNF induces the neural differentiation of hUCB-MSCs in culture via the TrkB-mediated phosphorylation of ERKs and β-catenin and following transplantation into the developing brain.
Keywords
Introduction
Stem cells are highly attractive candidates for use in the treatment of a wide variety of human diseases. Among the various stem cells, mesenchymal stem cells (MSCs) show particular potential for clinical use because they can be obtained and cultivated easily. In addition, they self-renew with a high proliferative capacity and can differentgiate into multiple lineages (9,20,48,59). Human umbilical cord blood-derived MSCs (hUCB-MSCs) have proven to be more advantageous than bone marrow-derived MSCs in terms of cell procurement, storage, and transplantation (28). Moreover, the number and differentiation ability of bone marrow-derived MSCs significantly decrease with age (43). These characteristics make hUCB-MSCs potent candidates for the clinical application of allogenic MSC-based therapies.
There have been many attempts to induce the trans-differentiation of MSCs into neural cells, which could be used as a replacement cell therapy for neurological diseases (19,56). MSCs can differentiate into neurons in vitro under special experimental conditions (7,27). However, artifacts created by in vitro chemical stress are common (40,46). For example, the neuron-like morphological and immunocytochemical changes observed in MSCs after induction with dimethyl sulfoxide/butylated hydroxyl anisole (DMSO/BHA) are not the result of genuine neurofilament extension, but rather represent cell shrinkage and cytoskeleton retraction in response to chemical stress (40).
A more recent study demonstrated that MSCs can be primed toward a neural fate by the constitutive expression of neuronal antigens in culture in the absence of specialized induction reagents (17). MSCs also seem to respond with an appropriate neural pattern of differentiation when exposed to the environment of the developing brain. Furthermore, MSCs transplanted into an ischemic region of the rat brain or into sites of spinal cord injury are capable of differentiation into neural cells and promote functional improvement (35,61). However, after transplantation, survival of the transplanted cells can be poor, with few cells retaining the capability to differentiate into neurons (8,41).
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family of proteins. BDNF regulates the survival, development, and function of neurons (25) and is important for axonal regeneration, synaptic formation (14,55), and differentiation of neural precursor cells (1). In addition, it is essential for the induction of long-term potentiation and is one of the cellular bases for learning and memory (30,31,47). Binding of BDNF to its receptor, tyrosine kinase B (TrkB), is important for brain function, as it leads to the activation of several signal pathways involved in neural differentiation, development, and survival (6,25).
The present study investigated the neurogenic potential of BDNF in converting the mesodermal cell fate of hUCB-MSCs into a neural fate in culture in the absence of specialized induction chemicals and following transplantation into the developing nervous system. The molecular mechanisms involved in the BDNF-regulated cellular differentiation process were also explored.
Materials and Methods
Plasmid Construction
BDNF cDNA was obtained using the Marathon cDNA Amplification System (Clontech, Palo Alto, CA) and was cloned in the TOPO TA cloning vector (Invitrogen, Carlsbad, CA). Sequence integrity was verified. The plasmid encoding human BDNF was generated via polymerase chain reaction (PCR) cloning into pEGFP-N1 using KpnI (5′ primers) and NotI (3′ primers) restriction sites.
hUCB-MSCs and Transfection
Human UCB samples were collected from the umbilical vein of deliveries with informed maternal consent. A 16-gauge needle of a UCB collection bag containing 44.8 ml of CPDA-1 anticoagulant (Greencross, Yongin, Korea) was inserted into the umbilical vein and UCB was collected by gravity. Isolation and expansion of UCB-MSCs were conducted as previously reported (57). Ethical approval for the use of hUCB-MSCs was obtained from the Institutional Review Board of Catholic University Medical Center.
Cells were transfected with the BDNF-expressing plasmid using the Neon™ transfection system (Invitrogen). DNA (2 μg) was mixed with resuspension buffer. Then, 2 × 105 cells were electroporated with a preoptimized square pulse condition (1600 V, 20 ms, one pulse). Electroporated cells were incubated in α-minimum essential medium (α-MEM; Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Invitrogen) without antibiotics for 24 h. The media were then replaced with antibiotic-containing medium.
Immunophenotyping of hUCB-MSCs
To analyze the cell surface expression of typical marker proteins in hUCB-MSCs or BDNF-overexpressing hUCB-MSCs (MSCs-BDNF), cells were labeled with the following anti-human antibodies: CD34-PE (phycoerythrin), CD44-PE, CD73-PE, and CD90-PE (BD Biosciences, San Jose, CA). Ten thousand cells were measured using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ) and the results were analyzed with CellQuest software (Becton Dickinson).
Multidifferentiation of Cultured hUCB-MSCs
Differentiation to adipogenic and osteogenic lineages was induced according to previously described procedures (48). After 2–3 weeks of culture, differentiated cells were fixed with 3% formaldehyde. Adipocytes were detected by staining the lipid droplets in the cell using 0.3% Oil red-O stain for 10 min. Osteocytes were detected by calcium phosphate deposits after von Kossa staining. In brief, cells were fixed with ethanol and stained with 5% silver nitrate for 1 h. After rinsing with distilled water, the cells were incubated in 5% sodium thiosulfate for 2 min to allow precipitation of insoluble black silver granules around the calcium phosphate.
Immunocytochemistry
After transfection, cells were fixed with 4% paraformaldehyde for 10 min and processed for immunocytochemistry to identify neural marker-positive cells. Nonspecific antibody reactions were blocked with 5% horse serum for 1 h at room temperature (RT). Fixed cells were then incubated overnight at 4°C with primary antibodies directed against BDNF (Chemicon, Temecula, CA), βIII tubulin (Chemicon), neuronal nuclei (NeuN; Chemicon), glial fibrillary acidic protein (GFAP; Dako, Carpinteria, CA), and myelin basic protein (MBP; Chemicon). Antibody reaction was visualized with Alexa Fluor 546-conjugated anti-mouse or anti-rabbit secondary antibodies (Molecular Probes, Eugene, OR). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO) for counterstaining. Fluorescent images were acquired using a LSM 510 Meta confocal microscope (Carl Zeiss, Jena, Germany).
Western Blot Analysis
Antibodies were obtained from commercial sources: BDNF, βIII tubulin, NeuN, and MBP antibodies from Chemicon; GFAP antibody from Dako; tubulin, β-catenin, and p-β-catenin antibodies from Abcam (Cambridge, UK); poly ADP ribose polymerase (PARP) antibody from BD Bioscience; and extracellular signal-related kinases (ERKs), p-ERKs, and p-Raf-1 antibodies from New England Biolabs (Ipswich, MA). For Western blot analysis, cells were rinsed with phosphate-buffered saline (PBS) and were subsequently lysed for 30 min on ice in RIPA-B buffer (0.5% Nonidet P-40, 20 mM Tris, pH 8.0, 50 mM NaCl, 50 mM NaF, 100 μM Na3VO4, 1 mM DTT, and 50 μg/ml PMSF). The insoluble material was removed by centrifugation at 12,000 rpm for 20 min at 4°C. The supernatant was then subjected to SDS-PAGE and Western blot analysis was performed. The blots were blocked in PBS with 5% skim milk and 0.05% Tween 20, incubated with the appropriate antibodies, and subsequently incubated with the secondary antibodies conjugated with horseradish peroxidase. The blots were then assayed using an enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ).
Subcellular Fractionation
To isolate nuclear, cytosol, and cytoskeleton fractions, cells were rinsed with PBS and were subsequently lysed using the Qproteome Cell Compartment Kit (Qiagen, Valencia, CA). The supernatant was confirmed by Western blot analysis using PARP as a nuclear marker, tubulin as a cytoskeleton marker, and ERKs as a cytoplasm marker.
Transplantation of MSCs Into the Neonatal Mouse Brain
All animals were handled in accordance with institutional guidelines under the approved protocols. hUCB-MSCs or MSCs-BDNF were labeled using a PKH26 Red Fluorescent Cell Linker kit (Sigma-Aldrich) according to the manufacturer's instructions. Briefly, cells were centrifuged for 5 min at 1,000 rpm and resuspended in 1 ml of the dilution buffer. The cell suspension was mixed with an equal volume of the labeling solution containing 4 × 10−6 M PKH26 in the dilution buffer and incubated for 5 min at room temperature. The reaction was terminated by adding 2 ml of FBS and cells were washed three times.
PKH26-labeled cells were transplanted into the lateral ventricle of postnatal day 1 ICR mice, as described previously (18). Briefly, newborn pups were anesthetized by hypothermia and placed in a clay mold. The head was transilluminated under a dissection microscope, and a Hamilton syringe with a beveled tip was lowered through the scalp and skull immediately anterior to the bregma. Approximately 1 × 105 cells in 2 μl of PBS were slowly injected into the left lateral ventricle (0.1 mm anterior to bregma, 0.8 mm lateral and 2.0 mm deep from the pial surface). Immediately after injection, pups were warmed in a 37°C incubator, and returned to the mother after approximately 30 min. After 7 days of survival, mice were perfused transcardially with 4% paraformaldehyde in PBS. The brain tissue was excised, postfixed overnight in perfusate, and then equilibrated in PBS containing 30% sucrose for 2 days. Fixed brains were embedded, snap frozen in liquid nitrogen, and stored at −70°C until use. Thirty mice in total were directly used to obtain the final data shown in this study: MSCs (n = 15) and MSCs-BDNF (n = 15).
Immunohistochemistry and Quantification
Tissues were cryosectioned at 14 μm in the coronal plane and then stained with primary antibodies for BDNF (Chemicon), NeuN (Chemicon), GFAP (Dako), and doublecortin (DCX; Chemicon) at 4°C overnight. The sections were incubated with Alexa 488-conjugated anti-IgG secondary antibodies (Molecular Probes) and counterstained with DAPI) (Sigma-Aldrich). Fluorescent images were acquired using a Zeiss LSM510 confocal microscope (Carl Zeiss).
To determine the numbers of the GFAP- and NeuN-positive cells, every 15th sagittal section (30 μm thick) was prepared from the forebrain in the hemisphere receiving cell engraftment. Total numbers of positive cells for the hemisphere were then obtained by multiplying by 3. All images were made using an excitation filter under reflected light fluorescence microscopy and transferred to a computer equipped with MetaMorph software version 7.5 (Molecular Devices, Downingtown, PA).
Results
Characterization of the BDNF-Mediated Neural Induction of MSCs
hUCB-MSCs stably overexpressing BDNF were obtained by transfecting hUCB-MSCs with a BDNF-expressing plasmid. MSCs-BDNF progressively assumed neuronal morphological characteristics over the course of the first day after transfection. The cytoplasm in the flat MSCs retracted toward the nucleus, resembling a typical neuronal soma, and the process became thinner because of continuous shrinkage of the cell body. The morphological changes progressed further over the subsequent 3 days to yield network-like structures and many long processes; these were sustained for more than 7 days. In contrast, control hUCB-MSCs exhibited a fibroblast-like morphology (Fig. 1A). In addition, the expression of MSC-related cell surface antigen was evaluated by flow cytometry. hUCB-MSCs exhibited a strong labeling for CD44, a receptor-III of extracellular matrix and CD73. CD73 has a distinct epitope on the membrane-bound ecto-5′-nucleotidase, which is a marker for src homology domains 3 and 4 (SH3 and SH4). hUCB-MSCs were strongly positive for CD90, and this indicated that they were in an undifferentiated state (5,57). However, labeling for these surface markers was decreased in MSCs-BDNF. The labeling for the hematopoietic cell marker CD34 was negative in both cell types (Fig. 1B). MSCs-BDNF also lost the potential to differentiate into adipocytes and osteocytes under conditions in which hUCB-MSCs easily differentiated into these mesodermal cell types (Fig. 1C). Importantly, MSCs-BDNF expressed several neural proteins after transfection, which included the differentiating neuron marker Tuj1, the mature neuron marker NeuN, GFAP (which is typical of astrocytes), and the oligodendrocyte marker MBP, under proliferating conditions. In contrast, expression levels of these proteins were weak in nontransfected control hUCB-MSCs (Fig. 1D). Similar results were obtained in Western blot experiments (Fig. 1E).

Characterization of brain-derived neurotrophic factor overexpressing mesenchymal stem cells (MSCs-BDNF). (A) Time-lapse, phase-contrast microscopy analysis of MSCs at different time points after transfection of MSCs with a BDNF-expressing plasmid. The MSCs-BDNF assumed a neuron-like morphology, which was sustained for more than 7 days. The results are representative of at least three independent experiments. Scale bar: 200 μm. (B) Immunophenotyping of MSCs before and 3 days after transfection of MSCs with a BDNF-expressing plasmid. Cells were labeled with antibodies against the indicated antigens and then analyzed by flow cytometry. The results are representative of at least three independent experiments. (C) MSCs were transfected with a BDNF-expressing plasmid. After transfection, cells were allowed to differentiate under the specific induction medium. The accumulation of calcium-containing mineral deposits in the extracellular matrix and lipidic vacuoles were revealed by the (upper panel) von Kossa staining method and (bottom panel) Oil Red O solution after 3 weeks. Scale bar:; 200 μm (upper level) and 100 μm (bottom panel). The results were representative of at least two independent experiments. (D) Immunostaining of MSCs before and 3 days after transfection of MSCs with a BDNF-expressing plasmid. Cells were fixed and stained with mouse antibodies against the neural lineage markers Tuj-1 (neuron-specific class III β-tubulin), NeuN (neuronal nuclei), GFAP (glial fibrillary acidic protein), and MBP (myelin basic protein). The MSCs-BDNF exhibited stronger labeling for these markers than did control MSCs. The results were representative of at least three independent experiments. Scale bar: 50 μm. (E) Western blotting of MSCs before and after 1, 3, and 7 days of transfection of MSCs with a BDNF-expressing plasmid. Cell lysates were then subjected to SDS-PAGE and Western blot analysis using anti-Tuj-1, anti-NeuN, anti-GFAP, and anti-MBP antibodies. Loading of equal amounts of total protein were confirmed using anti-β-actin antibody. The results shown here are derived from one of two independent experiments.
Both the β-Catenin and ERK Signals Are Involved in the BDNF-Stimulated and TrkB-Mediated Neural Differentiation of MSCs in Culture
Figure 1 depicts the effect of BDNF on the promotion of the differentiation of hUCB-MSCs into neural cells in culture. The next experiment investigated the molecular mechanisms that controled this phenomenon. A significant upregulation of the phosphorylation levels of TrkB, which is a receptor of BDNF, was observed (Fig. 2A). Interestingly, the phosphorylation levels of β-catenin Y654 were increased dramatically in hUCB-MSCs after transfection with BDNF (Fig. 2B). These results suggested that BDNF stimulates the upregulation of the phosphorylation of TrkB and β-catenin. To elucidate the involvement of the TrkB and β-catenin signals in BDNF-stimulated neural differentiation, the BDNF-expressing plasmid was transfected into hUCB-MSCs and the cells were treated with the TrkB-specific inhibitor K252a. Treatment with 100 nM K252a inhibited the phosphorylation of TrkB and β-catenin Y654 (Fig. 2C), which suggests that BDNF stimulates neural differentiation via TrkB-mediated β-catenin phosphorylation in hUCB-MSCs.

Increase in the phosphorylation level of TrkB and β-catenin in presumed neurons derived from MSCs-BDNF. (A, B) Western blot analysis of MSCs before and 3 days after transfection of MSCs with a BDNF-expressing plasmid. Cell lysates were then subjected to SDS-PAGE and Western blot analysis using anti-phospho-β-catenin, anti-β-catenin, anti-phospho-TrkB, and anti-TrkB antibodies. Loading of equal amounts of total protein were confirmed using an anti-β-actin antibody. The blots are representative of at least two independent experiments. (C) Cells were transfected with a BDNF-expressing plasmid for 3 days and were then treated with 100 nM of the TrkB inhibitor K252a for 16 h; the levels of protein expression and phosphorylation were examined using Western blot analysis. Equal amounts of total protein were confirmed by using an anti-β-actin antibody. The results shown here are from one of two independent experiments.
To elucidate the downstream events resulting from β-catenin tyrosine phosphorylation at Y654 the subcellular localization of phosphorylated forms Y654-P β-catenin was examined. In canonical Wnt signaling, nuclear β-catenin binds to the promoter of T-cell factor-lymphoid-enhancing-factor (TCF/LEF) to regulate gene transcription (11,39). Y654-P was detected in the cytoplasm and cytoskeleton, along the process in MSCs-BDNF, but not in nucleus (Fig. 3). The results supported the view that phosphorylation of β-catenin at Y654 by BDNF is locally regulated in the cytoskeleton by a TCF transcription-independent mechanism.

Localization of phosphorylated forms Y654-P β-catenin in presumed neurons derived from MSCs-BDNF. (A) Western blot analysis of MSCs 3 days after transfection of MSCs with a BDNF-expressing plasmid. Nuclear, cytoplasmic, and cytoskeleton fractions were prepared and subjected to SDS-PAGE, and the resolved proteins were analyzed by Western blot using anti-phospho-β-catenin, anti-ERKs (extracellular signal regulated kinases), anti-PARP (poly ADP ribose polymerase), and anti-tubulin antibodies. The blots are representative of at least three independent experiments. (B) Immunostaining of MSCs 3 days after transfection of MSCs with a BDNF-expressing plasmid. Cells were fixed and stained with mouse antibody against phospho-β-catenin immunolabeled with red fluorescence. Anti-Y654-P-β-catenin antibodies stained the cell cytoskeleton and cytoplasm, but the nucleus was negative. The results are representative of at least three independent experiments. Nuclei were counterstained with DAPI (blue). Scale bar: 50 μm.
We reported previously that ERK signals are activated after the neural induction of MSCs via the use of various factors or antioxidants (36,37). Presently, the phosphorylation levels of Raf-1 and ERKs were dramatically increased in MSCs-BDNF compared with control hUCB-MSCs (Fig. 4A). In addition, treatment of MSCs-BDNF with K252a abrogated ERK phosphorylation effectively (Fig. 4B). These results indicated that ERK signals may also be involved in the BDNF-stimulated and TrkB-mediated neural differentiation of hUCB-MSCs in culture.

Increase in the phosphorylation level of ERKs in presumed neurons derived from MSCs-BDNF. (A) Western blot analysis of MSCs before and 3 days after transfection of MSCs with a BDNF-expressing plasmid. Cell lysates were then subjected to SDS-PAGE and Western blot analysis using anti-phospho-Raf-1, anti-Raf-1, anti-phospho-ERKs, and anti-ERKs antibodies. Loading of equal amounts of total protein were confirmed using anti-β-actin antibody. The blots are representative of at least two independent experiments. (B) Cells were transfected with a BDNF-expressing plasmid for 3 days and were then treated with 100 nM of the TrkB inhibitor K252a for 16 h; the levels of protein expression and phosphorylation were examined using Western blot analysis. Loading of equal amounts of total protein were confirmed using anti-β-actin antibody. The blots are representative of at least two independent experiments.
Although both the β-catenin and ERK pathways are crucial for neural differentiation, no significant interactions between these signals have been identified. The cross-talk between the β-catenin and ERK pathways during the regulation of neural differentiation was analyzed using small interfering RNA (siRNA) transfection. As shown in Figure 5A, transfection with a β-catenin siRNA resulted in a significant decrease in the level of ERK phosphorylation in hUCB-MSCs treated with recombinant BDNF. In contrast, transfection of the same cells with an ERK siRNA had no effect on the levels of phosphorylation of β-catenin (Fig. 5B), which suggests that BDNF activates ERK signals via a mechanism that is dependent on the action of β-catenin as an upstream regulator. The next experiment further investigated the involvement of β-catenin and ERKs in BDNF-stimulated neural differentiation of hUCB-MSCs. Transfection with β-catenin siRNA abrogated the expression of neural proteins in hUCB-MSCs treated with recombinant BDNF (Fig. 6A). Similar results were obtained in ERK siRNA experiments (Fig. 6B). These results were consistent with the requirement for β-catenin and ERK for BDNF-stimulated neural differentiation of hUCB-MSCs.
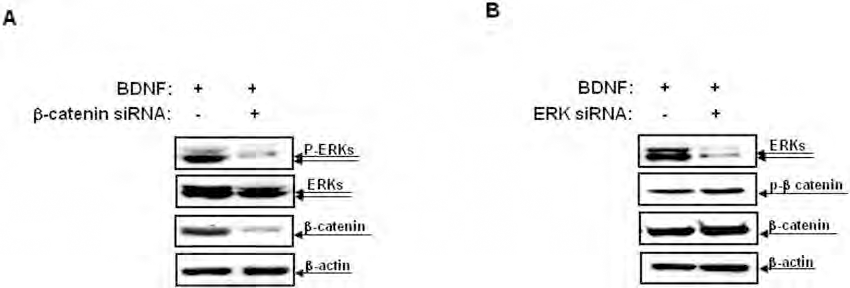
Cross-talk between the β-catenin and the ERK signals in presumed neurons derived from MSCs-BDNF. (A) MSCs were transfected with a β-catenin siRNA for 2 days and were then treated with 50 ng/ml of BDNF for 48 h. Cell lysates were then subjected to SDS-PAGE and Western blot analysis using anti-phospho-ERKs, anti-ERKs, and anti-β-catenin antibodies. Loading of equal amounts of total protein were confirmed using anti-β-actin antibody. The blots are representative of at least two independent experiments. (B) MSCs were transfected with an ERK siRNA for 2 days and were then treated with 50 ng/ml of BDNF for 48 h. Cell lysates were then subjected to SDS-PAGE and Western blot analysis using anti-phospho-β-catenin, anti-β-catenin, and anti-ERKs antibodies. Loading of equal amounts of total protein were confirmed using anti-β-actin antibody. The blots are representative of at least two independent experiments.

Effects of β-catenin and ERK in presumed neurons derived from MSCs-BDNF. (A) MSCs were transfected with a β-catenin siRNA or (B) an ERK siRNA for 2 days and were then treated with 50 ng/ml of BDNF for 48 h. Cell lysates were then subjected to SDS-PAGE and Western blot analysis using anti-Tuj-1, anti-NeuN, anti-GFAP, and anti-MBP antibodies. Loading of equal amounts of total protein were confirmed using an anti-β-actin antibody. The results shown here are derived from one of two independent experiments.
MSCs-BDNF Exhibited Neural Differentiation After Grafting Into the Neonatal Mouse Brain
To test the in vivo transdifferentiation capacity of MSCs-BDNF, PKH26-labeled MSCs or MSCs-BDNF were grafted into the lateral ventricles of neonatal mouse brains. PKH26 is incorporated as aliphatic reporter molecule into the cell membrane by selective partitioning (23). PKH26 has been useful for in vitro as well as in vivo cell tracking applications. It is especially favored for its low levels of toxicity and long half-life in in vivo studies (21,26,50).
Seven days after transplantation, a significant increase in the number of labeled cells was evident around the ventricular zone and within the olfactory bulb in MSCs-BDNF-transplanted mice compared with mice transplanted with hUCB-MSCs alone. In the MSCs-transplanted group, most of the labeled cells were near the site of injection within the subependymal zone of the lateral ventricle, with a small number of labeled cells detected within the olfactory bulb in mice transplanted with hUCB-MSCs alone (Fig. 7A, B). In addition, hUCB-MSCs transfected with the BDNF gene retained strong transgene expression in vivo (Fig. 7C).

Engraftment and distribution of transplanted cells in the neonatal mouse brain. PKH26-labeled MSCs or MSCs-BDNF (1 × 105 cells) were implanted into the lateral ventricle of the neonatal mouse brain (n = 7 per treatment group). (A) Many PKH26-positive (red) cells were observed around the ventricular zone of MSCs-BDNF-transplanted mice 7 days after transplantation (c and d, box from c). In the MSCs-alone group, most of PKH26-positive cells were observed near the site of injection within the subependymal zone of the lateral ventricle (a and b, box from a). (B) A significantly increased number of PKH26-positive cells were observed within the olfactory bulb of MSCs-BDNF-transplanted mice (c and d, box from c) compared with the MSCs-alone group 7 days after transplantation (a and b, box from a). (C) A subset of cells was stained for BDNF 7 days after transplantation with MSCs-BDNF. Nuclei were counterstained with DAPI (blue). Scale bar: 100 μm (a, c); 50 μm (b, d).
Some of the grafted PKH26-labeled cells penetrated into the overlaying parenchyma. Immunolabeling showed that some of these cells were positive for staining with the anti-GFAP antibody (Fig. 8A). In addition, a small subset of the grafted cells was immunopositive for NeuN within the olfactory bulb (Fig. 8B). Compared with hUCB-MSCs, transplantation of MSCs-BDNF further increased the number of GFAP-positive cells by 2.8-fold, and the number of NeuN-positive cells by 3.9fold.
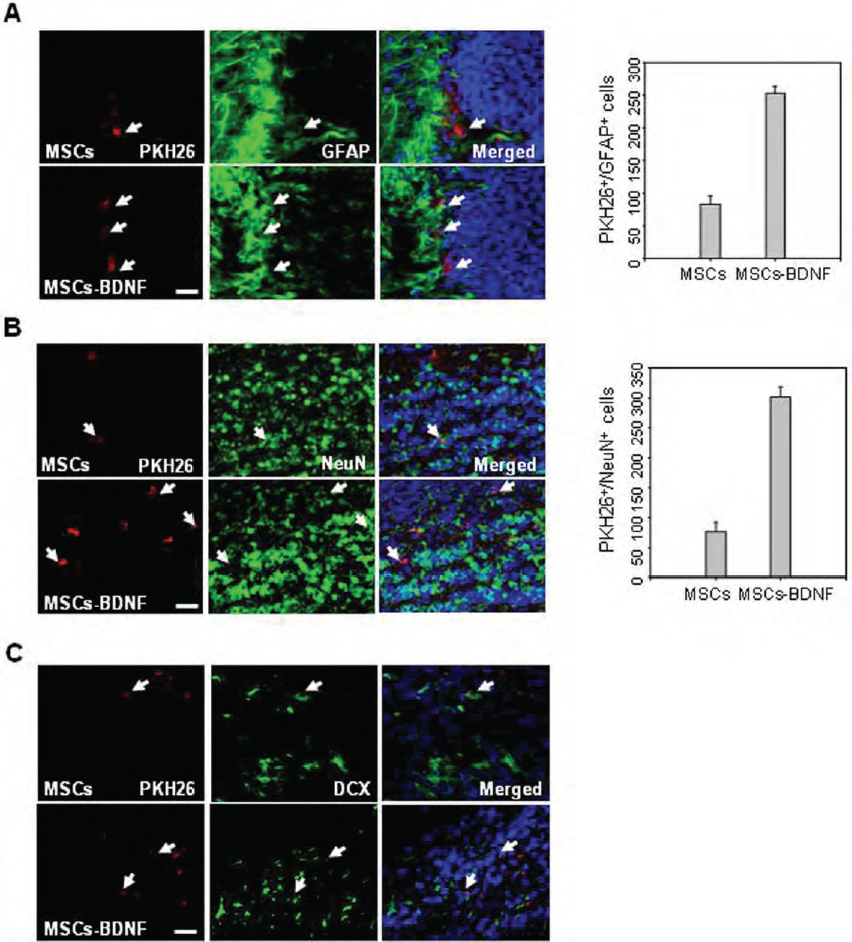
Differentiation of transplanted cells into neural-lineage cells in the neonatal mouse brain. A subset of the transplanted PKH26-labeled cells expressed (A) GFAP in the subventricular zone and (B) NeuN in the olfactory bulb 7 days after implantation (1 × 105 cells). PKH26-GFAP or PKH2-NeuN double-positive cells were counted. Data are presented as mean numbers of positive cells ± SD. (C) A subset of PKH26-labeled cells expressed doublecortin (DCX) in the ventricular zone 7 days after implantation (n = 8 per treatment group). These markers were immunolabeled with green fluorescence. Nuclei were counterstained with DAPI (blue). Scale bar: 50 μm.
Furthermore, immunolabeling revealed that some of transplanted PKH26-labeled cells were positive for DCX, a surface protein expressed on migratory neuroblasts, in MSCs and MSCs-BDNF-transplanted mice (Fig. 8C).
Discussion
MSCs are an attractive and valuable cell source for clinical applications because of their high proliferative capacity and their ability to differentiate into multiple lineages (9,20,48,59). Moreover, MSCs are not immunogenic; therefore, they do not elicit a proliferative response of allogeneic lymphocytes in vitro (33).
MSCs display some neural properties constitutively in culture and appear to differentiate into neural lineage cells when exposed to the environment of the developing nervous system (17). We showed previously that treatment of hUCB-MSCs with BDNF in neural induction medium facilitates their neural differentiation in vitro (37). Although this induction method using various antioxidants is used widely for the induction of rapid differentiation into neurons, it suffered from artifacts created by in vitro chemical stress (40,46). One of these studies chronicled the neuron-like morphological and immunocytochemical changes observed in MSCs after induction with DMSO/BHA are not the result of genuine neurofilament extension, but rather they represent cell shrinkage and cytoskeleton retraction in response to chemical stress (40). Another study reported a rapid and dramatic morphological change in NIH3T3, similar to that of MSCs upon dibutyryl cAMP/isobutylmethylxanthine (dbcAMP/IBMX) treatment without expression of neuronal marker (17).
The present study investigated the neurogenic potential of BDNF in converting hUCB-MSCs into a neural fate in culture in the absence of specialized induction chemicals reagents. BDNF transfection decreased the mesodermal cell fate and induced the neural characteristics of hUCB-MSCs in vitro (Fig. 1).
A diverse array of extracellular stimuli including growth factors and neurotrophins can trigger the activation of several intracellular pathways that play a major role in neuronal differentiation and synapse formation during the development of the nervous system (2,11,15,24,53). This can produce a cascade of responses that may lead to the functional and morphological alteration of neurons (22,51). Neurotrophins and their Trk receptors are among the best known regulators of neuronal development and function (10,24,25,60). In particular, TrkB is expressed in the hippocampus and its ligand, BDNF, regulates neurite outgrowth and branching in hippocampal neurons (13,32,38,42,55). BDNF is important for differentiation of neural precursor cells, axonal regeneration, and synaptic formation (1,14,55). BDNF directly enhances the differentiation of CNS stem cell-derived neuronal precursors (1).
What are the mechanisms that are implicated in the BDNF-regulated cellular differentiation process? Presently, BDNF stimulated hUCB-MSCs neural differentiation via the TrkB-mediated phosphorylation of β-catenin (Fig. 2). In the nervous system, β-catenin regulates axon extension (49), dendritogenesis (58), and synaptic assembly and plasticity (3,4,52,58). In addition, tyrosine phosphorylation of β-catenin regulates synapse formation and function (4,44).
β-Catenin is a cytoplasmic protein that plays a crucial role in Wnt signaling. In the canonical Wnt pathway, Wnt proteins inhibit glycogen synthase kinase-3β (GSK-3β), which normally targets β-catenin for degradation, resulting in β-catenin nuclear translocation and activation of TCF/LEF-driven gene transcription. Consequently, TCF factors are likely candidates in the response activated by nuclear β-catenin (11,39,45). The effects of BDNF on axonal growth and branching in hippocampal neuronal cells via the phosphorylation of β-catenin Y654 have been reported (16). Furthermore, β-catenin phosphorylated at Y654 is localized in the cytoskeleton at growth cones but not in the nucleus, which indicates that BDNF regulates axonal morphogenesis of hippocampal neuronal cells by TCF4-independent mechanisms. β-Catenin phosphorylated at Y654 also localized in the cytosol and cytoskeleton in MSCs-BDNF, which suggests that phosphorylation of β-catenin at Y654 locally occurs independent of TCF mechanisms during the BDNF-stimulated neural differentiation of hUCB-MSCs (Fig. 3). Further studies should consider the BDNF-mediated β-catenin downstream signaling in neural differentiation.
The Ras-ERK pathway is also a major signaling route involved in the regulation of neural differentiation (12,29,34,54). We reported previously that ERK signals are activated after the neural induction of MSCs (36,37). Presently, BDNF increased the expression levels of Raf-1 and ERK phosphorylation (Fig. 4). In addition, BDNF activated ERK signals in a manner that was dependent on β-catenin as an upstream regulator (Fig. 5). Although both the β-catenin and ERKs pathways are major signaling pathways involved in neural differentiation during the development of nervous system, no significant interactions between these pathways have been identified. Therefore, our results contribute to the clarification of the cross-talk between the β-catenin and ERK pathways in the regulation of the BDNF-stimulated neural differentiation process. Furthermore, the inhibition of β-catenin and ERK abrogated the BDNF-stimulated upregulation of neuronal phenotype markers (Fig. 6). These results demonstrate that β-catenin and ERK are required for BDNF-stimulated neural differentiation of hUCB-MSCs.
Although the neural differentiation capacity of MSCs in vitro has been explored widely, the in vivo response of this cell type after transplantation into the brain has not been assessed adequately. The current finding that an increased number of cells engrafted into the developing brain differentiated significantly into neurons in the olfactory bulb and periventricular astrocytes in MSCs-BDNF-transplanted mice compared with mice transplanted with hUCB-MSCs alone (Fig. 7) supports the conclusion that BDNF induces the neural differentiation of hUCB-MSCs in vitro. These results may indicate a potential therapeutic use for BDNF-expressing MSCs as neural progenitor cells for the replacement of lost neural tissue after injury.
In conclusion, BDNF functions as an effective stimulator in converting the hUCB-MSCs into a neural fate in culture in the absence of specialized induction chemicals via the TrkB-mediated phosphorylation of ERKs and β-catenin phosphorylation and following transplantation into the developing brain. Further work using various models of neurological disease (designed to assess fully the ability of MSCs induced by BDNF into a neuronal fate to functionally integrate into the neural circuitry) will determine the potential therapeutic value of these cells in the treatment of neurological injury and disease.
Footnotes
Acknowledgments
This study was supported by a grant of the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea (0405-DB01-0104-0006), and by Basic Science Research program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2010-0017243 and 2010-0022845), Republic of Korea. The authors declare no conflicts of interest.