
Abstract
Long-term engraftment and phenotype correction has been difficult to achieve in humans after in utero stem cell transplantation mainly because of allogeneic rejection. Autologous cells could be obtained during gestation from the amniotic fluid with minimal risk for the fetus and the mother. Using a sheep model, we explored the possibility of using amniotic fluid mesenchymal stem cells (AFMSCs) for autologous in utero stem cell/gene therapy. We collected amniotic fluid (AF) under ultrasound-guided amniocentesis in early gestation pregnant sheep (n = 9, 58 days of gestation, term = 145 days). AFMSCs were isolated and expanded in all sampled fetal sheep. Those cells were transduced using an HIV vector encoding enhanced green fluorescent protein (GFP) with 63.2% (range 38.3–96.2%) transduction efficiency rate. After expansion, transduced AFMSCs were injected into the peritoneal cavity of each donor fetal sheep at 76 days under ultrasound guidance. One ewe miscarried twin fetuses after amniocentesis. Intraperitoneal injection was successful in the remaining 7 fetal sheep giving a 78% survival for the full procedure. Tissues were sampled at postmortem examination 2 weeks later. PCR analysis detected GFP-positive cells in fetal tissues including liver, heart, placenta, membrane, umbilical cord, adrenal gland, and muscle. GFP protein was detected in these tissues by Western blotting and further confirmed by cytofluorimetric and immunofluorescence analyses. This is the first demonstration of autologous stem cell transplantation in the fetus using AFMSCs. Autologous cells derived from AF showed widespread organ migration and could offer an alternative way to ameliorate prenatal congenital disease.
Introduction
Many congenital diseases have a relatively low prevalence, but collectively they represent a large burden of disease (28). It is estimated that they are responsible for over a third of all pediatric hospital admissions, and for up to 50% of the total cost of pediatric hospital treatment (28). Due to the improvement in medical care, affected patients can now survive to adulthood, requiring continued therapy.
In utero stem cell transplantation (IUSCT) aims to treat congenital disorders in the fetus using cells capable of self-renewal that can enhance or substitute the affected tissues/organs of the fetus. The small fetal size provides a stoichiometric advantage, allowing the transplantation of much larger cell doses on a per kilogram basis than can be achieved after birth (4,7). Prenatal treatment could also avoid complications occurring in the postnatal period, as some genetic diseases damage the organ in utero and lead to irreversible changes (10). Finally, the immunological naivety in the early gestation fetus has given rise to the concept of fetal tolerance, the inability to raise an immunological response against foreign antigens (21). However, it is now clear that the human fetus may have a functionally developed immune system as early as the second trimester of gestation (49) and allogeneic cells injected at that time engraft poorly (30,35). In humans, successful IUSCT using allogeneic hematopoietic stem cells (HSCs) has been limited to fetuses with severe immunologic defects, where the effective lack of immune response to allogeneic cells mean that transplanted genetically normal cells have a proliferative advantage (46).
Mesenchymal stem cells (MSCs) appear to be less immunogenic than their hematopoietic counterparts (2,34). IUSCT of first trimester human fetal blood MSCs in mice with osteogenesis imperfecta (OI) was reported to ameliorate skeletal pathology (23). In a human fetus affected by OI successful engraftment of allogeneic MSCs with 5% chimerism was obtained at 8 months of age (27). In normal fetal sheep, where the immune system is believed to develop from 65 to 70 days of gestation (29), transplantation of allogeneic fetal liver cells resulted in levels of mixed chimerism as high as 30% after a single intrauterine dose (19,54,55) and low-level engraftment lasted for over 2 years. Increasing the numbers of T-cells added back to the transplant increased the level of chimerism, although the corresponding increased risk of graft-versus-host disease limits the number of allogeneic cells that can be transplanted (6,8). Attempts to treat other diseases such as sickle cell or metabolic storage disorders have been unsuccessful, even where a suitably matched donor has been available (14,51).
Autologous cells could potentially overcome these obstacles (1,35,38,43,53). Multi- and pluripotent stem cells can be easily derived from amniotic fluid (5,15) and their ability to differentiate into various lineages such as myogenic, adipogenic, osteogenic, neuronal, endothelial, and hepatic cells has been extensively investigated (15,16,24–26,48). Moreover, we and others have demonstrated that amniotic fluid-derived stem cells can be easily transduced without losing their potential (16,22). Therefore, transplantation of genetically corrected autologous amniotic fluid stem cells into a fetus could represent an alternative therapeutic strategy for the treatment of severe congenital diseases.
In this study, we investigated the feasibility and potential for amniotic fluid mesenchymal stem cells (AFMSCs) in order to combine IUSCT and gene therapy into one therapeutic strategy. With this aim we used the fetal sheep, which has the advantages of long gestational age, similar fetal size, and comparable fetal development and physiology to human pregnancy (10,13) and in which there is wide experience with fetal interventional procedures (9,12).
Materials and Methods
Animal Procedures
Nine fetuses from seven ewes (Romney breed) were used in these experiments. The experimental design is illustrated in Figure 1A. Ewes were time mated after receiving intravaginal progesterone suppositories for 2 weeks to induce ovulation. General anesthesia was induced with ketamine (4 mg/kg, Merial, UK) intravenously and, after intubation, the ewes were maintained on isoflurane-Vet 3% (Merial, UK) in oxygen. Fetal measurements were confirmed by ultrasound as previously described (11). The first procedure, amniocentesis (Fig. 1B), was performed in early pregnancy (57–61 days of gestation, term =145 days) under ultrasound guidance using a 22-gauge, 15-cm echo-tip needle (Cook Medical, USA) and 10 ml of amniotic fluid was withdrawn from each amniotic sac. After recovery fetal well-being was confirmed using ultrasound examination.

Experimental design and injection procedure. After amniocentesis collection, amniotic fluid mesenchymal stem cells (AFMSCs) were cultured in adherence in defined conditions (A). Cells were transfected with lentivirus GFP and reinjected into the peritoneal cavity of the fetal donor. Sonograms showing ultrasound guided amniocentesis (B) and intraperitoneal injection (C). Amniocentesis was performed using a 22-gauge needle to collect 10 ml amniotic fluid from the amniotic cavity around a fetal sheep at 58 days of gestation. For intraperitoneal injection of transduced expanded amniotic fluid cells we used a 20-gauge needle inserted through the anterior abdominal wall of a sheep fetus at 76 days of gestation. Echogenicity can be seen throughout the peritoneal cavity after injection of cells. Scale bars: 5 cm. (D) The timeline of the experiment.
Approximately 2 weeks later (71–83 days of gestation) sheep were reanesthetized as above for the second procedure. Transduced AFMSCs (1–2 ml) were injected under ultrasound guidance into the peritoneal cavity of the fetus from which they were derived (Fig. 1C) using a 20-gauge 15-cm echo-tip disposable needle (Cook Medical). To ensure correct needle placement within the peritoneal cavity, the needle was inserted through the anterior abdominal wall of the fetal sheep superior and lateral to the fetal bladder to avoid the umbilical arteries, and microbubbles were observed moving within the peritoneal cavity as the vector was instilled. Fetal well-being was examined after the procedure. A scheduled postmortem examination was performed approximately 3 weeks after intraperitoneal AFMSC transplantation (89–103 days of gestation). There was widespread sampling of maternal and fetal tissues that were fixed in 4% paraformaldehyde (pH 7.4) overnight and processed into wax for immunohistochemistry analysis. In parallel, tissues were also snap-frozen for protein extraction and PCR analysis. Cord blood, spleen, liver, and bone marrow of fetal sheep and maternal peripheral blood and bone marrow were collected for FACS flow cytometry. All procedures on animals were conducted in accordance with UK Home Office regulations and Guidance for the Operation of Animals (Scientific Procedures) Act (1986).
Ex Vivo Expansion of Amniotic Fluid Mesenchymal Stem Cells
Amniotic fluid samples collected from fetal sheep were centrifuged (300 × g) for 5 min. Pellets were resuspended in the amniotic culture medium (ACM), which consisted of aMEM (63%, Life Technology, Gaithersburg, MD), Chang medium (20%, Chang B plus Chang C; Irvine Scientific, Santa Ana, CA), fetal bovine serum (FBS, 15%, Invitrogen, UK), and streptomycin, penicillin, and L-glutamine (1% each, Invitrogen). Cell seeding was performed on single 35-mm Falcon petri dishes (Becton Dickinson, Los Angeles, CA) that incubated at 37°C with 95% air and 5% carbon dioxide. After 3 days, nonadherent cells and debris were discharged while changing new medium and the adherent cells were cultured until preconfluence. Adherent cells were detached from the plastic substrate using 0.05% trypsin and 0.02% sodium-EDTA (Life Tech). Cells were passaged into 100-mm petri dishes and further split one to four once the dish reached 70% confluence. Cell size and number was determined using a hemocytometer (Marienfeld, Germany) before and after viral transduction. The formula used for the doubling time (hours) was h*log(2)/log(c2/c1), where c1 is initial seeding cell number, c2 is cell number after growth, and h is hours of culture.
Cell Cycle Analysis
Propidium iodine (PI) was used to stain cell nucleic acid to identify the proportion of cells that are in one of the three interphase stages of the cell cycle. Briefly, 1 × 106 AFMSCs were washed in PBS and centrifuged to produce a cell pellet. Seventy percent ethanol (1 ml) was added to fix the cell pellet during centrifugation. After 30-min incubation on ice, cells were pelleted using a high-speed centrifuge at 3000 rpm for 5 min. Ribonuclease A solution (50 μl, 100 μg/ml) and PI solution (400 μl, 50 μg/ml) were added directly into the PBS washed pellet for 10 min at room temperature. The samples were analyzed by flow cytometry as described below.
Differentiation and Characterization of Amniotic Fluid Mesenchymal Stem Cells
In order to characterize the isolated, cultured, and transduced amniotic fluid cells, the following surface markers were used. Mouse anti-sheep CD14, CD31, CD44, CD45, and CD58 antibodies conjugated with fluorescein isothiocyanate (FITC) (AbD Serotec, UK), and mouse anti-human CD166 conjugated with phycoerythrin (PE) (BD Pharmingen, UK) were used for characterization of the nontransduced AFMSCs. Goat anti-mouse Alexa Fluor® 568 (Invitrogen) was used as secondary antibody to mouse CD14, CD31, CD44, CD45, and CD58 in order to identify the GFP-transduced AFMSC cells. Antibodies (10 μl) were stained with 5 × 105 cells in 100 μl ice-cold PBS. The incubation time was 15 min. The stained cells were analyzed with flow cytometry.
To induce adipogenic differentiation, cells from passage 2 were seeded at a density of 3,000 cells/cm2 and were cultured for 21 days in DMEM low-glucose medium with 10% FBS, antibiotics (Pen/Strep, Gibco/BRL), and adipogenic supplements [1 mM dexamethasone, 1 mM 3-isobutyl-1-methylxanthine, 10 mg/ml insulin, 60 mM indomethacin (Sigma-Aldrich, UK)]. The cells were stained with Oil red O (Sigma-Aldrich). ACM was used as a control growth medium in control conditions.
For osteogenic differentiation, cells from passage 2 were seeded at a density of 3,000 cells/cm2 and were cultured for 21 days in DMEM low-glucose medium with 10% FBS (Gibco/BRL), Pen/Strep, and osteogenic supplements [100 nM dexamethasone, 10 mM betaglycerophosphate (Sigma-Aldrich), 0.05 mM ascorbic acid-2-phosphate (Wako Chemicals, UK)]. An alkaline phosphatase kit was used for osteogenic cells staining according to the manufacturer's instruction (Sigma-Aldrich). ACM was used as a control growth medium in control conditions.
Lentiviral Vectors Preparation and Transduction of Sheep Amniotic Fluid Stem Cells
A lentivirus vector encoding the the HIV-1 central polypurine tract element, the Spleen Focus Forming Virus LTR promoter, and the marker gene eGFP was used. Vector stocks were generated as previously described (17,39), and were concentrated by ultracentrifugation at 23,000 × g for 2 h at 4°C using a Beckmann ultracentrifuge. The virus particles were resuspended in X-Vivo 10 and stored at −80°C. The number of infectious particles was estimated by flow cytometry of 293T cells, 72 h after exposure to serial dilutions of virus stock. Cells cultured in ACM (1 × 106) and were seeded into 24-well non-tissue culture dishes. After 24 h they were transduced with the lentivirus vector at a multiplicity of infection of 10 virus particles per cell. Cells were incubated with the vector for 48 h at 37°C in 95% air and 5% carbon dioxide. Transduced cells were treated with trypsin and replated into 100-mm dishes for further growth in ACM.
Flow Cytometry Analysis
Single cell suspensions of fetal spleen, liver, and bone marrow were prepared immediately after animal sacrifice by straining fetal tissue through a 40-μm nylon mesh cell strainer (BD, UK). The fetal tissue was passed through the cell strainer with PBS rinsing. After washing with PBS, red blood cell lysis buffer was added to the cell pellet for 5 min at 37°C, which was then resuspended in PBS for flow cytometry analysis. Mononuclear cells were isolated from umbilical cord and maternal blood samples by density gradient centrifugation using Ficoll-Paque solution (Stem Cell Technology, Canada). Diluted blood solution (10 ml of blood and 25 ml of buffer) was centrifuged with Ficoll solution (15 ml) 30 min at 2000 rpm at room temperature without breaking to form the interphase. Then the interphase was carefully transferred to another new tube for further analysis. Single cell suspensions from control untransfected ewes and their fetuses were used as negative controls. The cells were acquired by Becton Dickinson FACSCalibur and LSR II machines (Becton Dickinson, San Jose, CA) and analyzed using FlowJo version 5.7.1 software (Tree Star). Ten thousand events were collected per sample and the data were stored as list mode files.
PCR Analysis
DNA was extracted from fetal and maternal tissues using a minikit (Invitrogen, USA) and the quality was confirmed using a nanodrop machine (NanoDrop Technologies, USA). Sample DNA (4 μg) was used for the first PCR reaction (30 cycles of 45 s at 65°C). The following primers were used for first round amplification of eGFP: forward, 5′-TGAACCGCATCGAGCTGAAGGG-3′ and reverse, 5′-TCCAGCAGGACCATGTGATCGC-3′. A second nested PCR round was performed (25 cycles of 45 s at 60°C) using the following second round primers: forward, 5′-GGCACAAGCTGGAGTACAACT-3′ and reverse, 5′-CCATGTGATCGCGCTTCT-3′. PCR products were analyzed on 1.2% agarose gel stained with ethidium bromide. DNA from transduced amniotic fluid cells was used as a positive control.
Western Blot Analysis
Cell pellets and homogenized tissues were resuspended in 200 μl PBS and 200 μl 2× protein sample buffer [0.5 M Tris-HCl (pH 6.8), 5% glycerol, 2% sodium dodecyl sulfate, and 100 mM dithiothreitol]. Samples were boiled for 5 min and centrifuged at 16,000 × g for 15 min to remove insoluble material. Samples were loaded onto a precast 7.5% Ready gel with a 4.5% stacker (BioRad Laboratories, Hercules, CA) and run for 1 h at 200 V. After electrophoresis, proteins were transferred to nitrocellulose membranes in a Bio-Rad Electroblotter Apparatus. Membranes were blocked in TBST (TBS, 0.05% Tween 20, Sigma, UK) containing 5% milk for 1 h at room temperature and then reacted overnight at 4°C with rabbit anti-GFP (Invitrogen, USA) diluted 1:2000 in TBST-milk. After three washes in TBST, the membranes were treated with affinity-purified horseradish peroxidase-conjugated goat anti-rabbit IgG (Promega, Madison, WI, USA) diluted 1:4000. Membranes were washed three times in TBST, and then developed with a 3,3′-diaminobenzidine-based horseradish peroxidase detection kit (Vector Laboratories, USA).
Immunofluorescence
Fresh fetal tissues were embedded in OCT, snap frozen in methyl-butane and liquid nitrogen, and cut into 10–15-μm sections using a cryostat (OTF, Bright, UK). Blocking solution was prepared with 1% BSA, 0.15% glycine, and 0.1% Triton in PBS and preserved at 4°C. To co-stain hepatocytes expressing GFP, mouse anti-sheep CK18 (Abcam, UK), mouse anti-sheep α-feto-protein antibody-2 (AFP) (Thermo Scientific, UK), and rabbit anti-GFP polyclonal (Invitrogen, USA) primary antibodies were applied at 1:150 concentration in blocking solution overnight at 4°C. Goat anti-rabbit (Alexa Fluor 488, Invitrogen, USA) and goat anti-mouse (Alexa Fluor 568) secondary antibodies were incubated for 2 h at room temperature with blocking solution at 1:150 concentration. To co-stain myocytes expressing GFP, rabbit anti-laminin (Abcam, UK) and mouse anti-GFP primary antibodes were applied. Secondary goat anti-rabbit (Alexa Fluor 594) and goat anti-mouse (Alexa Fluor 488) antibodies were used. The conditions for primary and secondary antibody staining were the same as for hepatocyte analysis. The stained tissues were covered by VECTASHIELD HardSet Mounting Medium with DAPI for nuclear staining (Vector Laboratories, USA). The slides were observed under inverted immunofluorescence microscopy (Leica, Germany). In order to quantify the number of AFMSCs migrated in the fetal liver, we counted GFP and CK18 positive/negative stained cells in four high-power fields per fetal liver. Values (mean ± SD) were expressed as the percentage of single- or double-positive stained cells per total nuclei in each high power field.
Results
Ultrasound Guided Amniocentesis and Intraperitoneal Injection of AFMSCs
Amniotic fluid was successfully sampled in all nine fetuses. One ewe subsequently miscarried twin fetal sheep a few days after the amniocentesis procedures, but before intraperitoneal injection could be performed. Intraperitoneal injection (n = 7) was successfully performed with no fetal or maternal mortality until planned postmortem examination in the remaining seven fetal sheep, giving a 78% survival for the full procedure. Experimental details are listed in Table 1. There was no maternal morbidity including fever, abdominal distension, hemorrhage, or ruptured amniotic membranes. There was no evidence of fetal hemorrhage or trauma relative to the injection procedure.
Experimental Details

After sampling of amniotic fluid (amniocentesis), cells were cultured and transduced in vitro, before being injected into the peritoneal cavity of the donor fetus (transplantation). Scheduled postmortem examination was performed approximately 3 weeks later to allow time for cell migration. nt: not tested.
Sheep AFMSC Isolation, Expansion, and Characterization
AFMSCs could be isolated and expanded in vitro from all the nine sheep (100%). The initial numbers of viable cells from 10 ml amniotic fluid in all animals were less than 20,000 cells, but they were easy to expand in petri dishes without feeder layer and demonstrated a doubling time ranging from 36 to 48 h (Fig. 2A). AFMSC growth velocity and differentiation potential was maintained for up to 10 passages (Fig. 2A). In order to achieve a sufficient number of cells for transplantation, cells were cultured for up to 20 passages. The number of injected cells was correlated with the time available for their culture from amniocentesis to transplantation (time of cell culture and transduction in days in Table 1). Subconfluent cells also showed no evidence of spontaneous differentiation (data not shown). The cell size and DNA content from cell cycle analysis did not show a significant difference between pre- and posttransduction AFMSCs (Fig. 2B, C). Samples of AFMSCs and GFP-AFMSCs had higher ratio in G0/G1 phase, with low percentage of S phase and G2/M phase (Fig. 2C).

Characterization of sheep amniotic fluid mesenchymal stem cells. The growth curve (A) demonstrates that the doubling time of both AFMSCs and GFP-FAMSCs was around 36–48 h (error bar: SD). (B) The cell size from AFMSCs and GFP-AFMSCs, which did not present statistical significance. (C) The DNA content of cells cycle by using flow cytometry analysis. The cell cycle did not showed significant difference between AFMSCs and GFP-AFMSCs. (D) The sheep AF cells stained strongly positive for MSC surface markers CD44, CD58, and CD166, confirming their mesenchymal origin. Anti-sheep CD14, CD31, and CD45 were negative compared to background staining (light gray, negative control; dark gray, anti-sheep antibodies).
The surface markers of mesenchymal stem cells, anti-sheep CD44, CD58, and CD166, were strongly positive in cultured sheep AF cells. The monocyte and lymphocyte markers, anti-sheep CD14, CD31, and CD45, showed negative in sheep cells (Fig. 2D). The results are the same either in the pre- or posttransduced AFMSCs. The two groups of cells that could be expanded in vitro showed the ability to differentiate into adipogenic and osteogenic lineages following the differentiation protocols (Fig. 3).

Differentiation potential of sheep amniotic fluid mesenchymal stem cells. Adipogenesis differentiation showing lipid drops with positive staining of Oil red O (A and insets) present in AFMSCs and GFP-AFMSCs. Moreover, both groups of cells could undergo osteogenic differentiation (B), expressed alkaline phosphatase, and formed lamellar-like structures (insets). Both AFMSCs and GFP-AFMSCs cultured in control conditions did not show adipogenic (A) and osteogenic differentiation (B). Scale bars: 20 μm.
AFMSCs Transduction with Lentivirus GFP Vector
Transduction efficiency, assessed 3 days after lentiviral transduction using cytofluorimetric analysis, was on average 63.2% (range 38.3–96.2%) (Fig. 4A, B, Table 1). GFP expression was also detected when AFMSCs were observed under fluorescence microscopy (Fig. 4C). Cell kinetics of GFP transduced AFMSCs was similar to nontransduced AFMSCs (Fig. 2A).
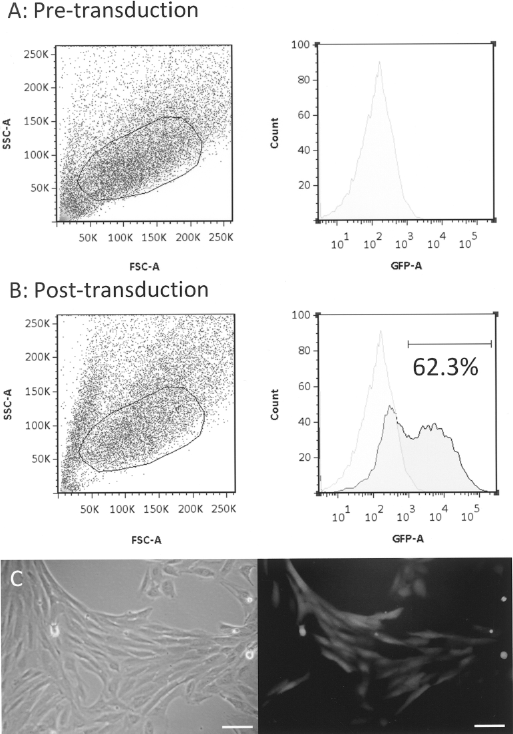
Transduction efficiency of AFMSCs. After transduction with lentivirus vector, AFMSCs were analyzed at the cytofluorimeter for GFP expression to determine the efficiency of gene transfer. For fetus 6, posttransduction cells in the GFP channel can be seen with 62.3% cells expressing GFP (B); the negative control is indicated (A). Cells were gated on the basis of the region shown on the forward (FSC-A) and side (SSC-A) scatter dot plot (left); the percentage of donor cells calculated by selection of the high fluorescence population on the cell count versus fluorescence intensity (GFP-A) histogram (right). AFMSCs were observed in bright field (C, left panel) and under a fluorescent microscope where the positive GFP expression could be detected within the cytoplasm of each AFMSC (C, right panel). Scale bars: 20 μm.
Detection of GFP DNA in Fetal and Maternal Organs
GFP DNA was detected in many fetal organs using nested PCR, suggesting systemic spread of GFP-positive cells after fetal injection (Table 2). The amniotic membrane and placentomes were most frequently positive (85.7%), followed by the fetal liver, which was positive in five fetuses (71.4%). GFP DNA was also detected in fetal muscle, umbilical cord, heart, adrenal gland, gonad, bone marrow, and peritoneum. The cell number we injected in each animal was varied. The length of cell culturing and final cell number for injection showed moderate correlation (r = 0.59) (Fig. 5A). There was no significant correlation between the number of injected cells and the presence of GFP-positive cells in the different organs as analyzed by PCR or Western blot [r = −0.18 (Fig. 5B) and r = −0.4 (Fig. 5C)]. Maternal liver, bone marrow, kidney, lung, heart, and adrenal gland were examined using GFP nested PCR but were negative, indicating no evidence of maternal engraftment after fetal transplantation (Fig. 5D).

The correlation between the number of injected cells and presence of GFP-positive cells in fetal organs, and evidence of maternal spread of GFP-AFMSCs. There was a moderate correlation (r = 0.59) between the final cell number and days of cell expansion in vitro (A). The correlation between the number of injected cells and the number of detectable GFP-positive organs did not showed significance in PCR (r = −0.18) (B) and Western blot (r = −0.4) (C). The PCR gel (D) showed all maternal organs analyzed, including adrenal gland, liver, lung, kidney, bone marrow (BM), and heart, were negative for GFP DNA detection. The fetal liver was strong positive. P, positive control; N, negative control.
PCR Analysis of GFP DNA and Western Blot of GFP Protein Expression in Seven Sheep Fetuses

Analysis of GFP Protein Expression in Fetal Organs
Cytofluorimetric analysis showed low-level presence of transduced AFMSCs in fetal bone marrow (0.85 ± 0.13%) and liver (1.24 ± 0.08%). Figure 6 shows one of the animals with positive stainin in bone marrow and liver. There were no GFP-positive cells in fetal umbilical cord blood and spleen, or maternal blood and bone marrow. GFP protein expression was also assessed by Western blot in those fetal tissues in which GFP DNA was detected (Table 2). There was insufficient protein available in samples of fetal gonad and peritoneum for Western blot analysis. The most frequent tissues to have positive GFP protein expression were the placenta (6/7, 86%), the fetal liver, and the amniotic membranes (5/7 fetuses, 71%, Table 2). Of all tissue samples that were PCR positive for GFP, Western blot confirmed GFP protein in 77.8% (21/27) of them. The presence of GFP protein detected 3 weeks after injection of transduced AFMSCs was strong evidence of cell migration in these tissues after transplantation.

Flow cytometric analysis for GFP expression in fetal blood, tissues, and maternal samples taken at postmortem examination. Histograms demonstrate positive GFP detection in the fetal liver (1.22%) and fetal bone marrow (0.84%). Cells from a control untransduced fetal sheep were used as negative control (light gray). Fetal cord blood, fetal spleen, maternal blood, and maternal bone marrow were negative for migration. BM: bone marrow.
Finally, to confirm the presence of GFP-positive AFMSCs in fetal tissue, we used immunofluorescence to analyze selective organs including fetal liver, skeletal and cardiac muscle, and placenta. For hepatocyte differentiation, we analyzed expression of GFP, cytokeratin 18 (CK18), and AFP in the liver of the four fetuses that had GFP-positive staining. Two liver lobes were examined in each fetus. In some sections, GFP-positive cells did not co-stain with CK18 (Fig. 7A–D). Only a few GFP-positive cells presented in animals were seen co-stained with CK18-positive cells in both liver lobes, suggestive of hepatocyte differentiation (Fig. 7E-G). Moreover, the percentages of positive cells per slide were 2.97 ± 1.15% for GFP+/CK18– cells and 1.08 ± 0.33% for GFP+/CK18+ cells in the fetal liver (Fig. 7H). For muscle differentiation, we analyzed the expression of GFP and laminin. GFP-positive cells were clearly detected in the skeletal muscle (Fig. 7I) and heart (Fig. 7J) of four fetuses. No myogenic differentiation could be observed when muscles were co-stained with laminin. Abundant GFP-positive cells could be detected in placentome (Fig. 7K–M).

Immunofluorescence for GFP expression and markers of MSC differentiation. GFP-positive AFMSC migration and differentiation was studied using GFP immunofluorescence and co-staining with tissue markers in fetal organs such as the liver (CK18; A–G) and skeletal muscle and heart (laminin; H–I). In fetal liver, GFP-positive cells (green) mainly localized in the stroma (A–D) and did not co-stain with CK18, a marker of hepatocytes stained red. In another section, however, a few rare cells co-stained for GFP and CK-18 (E–G, indicated by the arrowhead). Some cells that only stained for GFP remained in the stroma (F, indicated by the arrow). Histogram showed percentages of positive GFP cells with/without CK18 staining detected per high-power field in the liver section (H). Error bar: SD. In sections of fetal skeletal muscle (I) and heart (J), GFP-positive cells did not co-stain for laminin (red). Immunofluorescence of placental sections showed GFP-positive cells (K–M). Scale bars: 20 μm.
Both AFMSCs and GFP-AFMSCs did not show CK18 expression from our study, while the noninjected fetal sheep as a positive control showed strong expression of CK18 in the liver (Fig. 8A). AFP, another liver-specific marker, also demonstrated the presence of GFP-AFMSCs in the liver after autologous transplantation (Fig. 8B).

Immunofluorescence for CK18 and AFP expression in fetal liver and amniotic fluid stem cells. Upper panel shows positive CK18 expression in cells cultured from a control fetal sheep liver but no expression in amniotic fluid mesenchymal stem cells (AFMSCs) or transduced cells (GFP-AFMSCs) before injection. An uninjected sheep fetus of comparable gestational age was used as the control. The lower panel shows coexpression of GFP with expression of α-fetoprotein (AFP), another liver-specific marker in the fetal liver after transplantation of transduced AFMSCs. Scale bars: 20 μm.
Discussion
This is the first study to describe in utero autologous transplantation and migration of AFMSCs. We showed that using this system, it is possible to obtain a widespread migration of transduced autologous cells, with evidence of expression of transgenic protein in major fetal organs, bone marrow, and blood. Tissue analysis by PCR, Western blot, immunofluorescence, and cytofluorimetric assay revealed that AFMSCs injected into the peritoneal cavity preferentially localized in fetal liver, muscle, and heart. Moreover, a small number of cells expressing transgenic protein in the liver co-stained with markers of hepatocyte differentiation.
Using ultrasound-guided amniocentesis, a common clinical procedure with a known fetal loss rate of approximately 1% (45), we were able to isolate AFMSCs in 100% of the animals. A miscarriage occurred in one ewe carrying twin pregnancies despite a straightforward ultrasound-guided amniocentesis procedure. This compares favorably to the higher rates of fetal loss observed in other studies where different sources of autologous MSCs were used for autologous in utero transplantation. In particular, fetal transplantation of autologous MSCs derived from fetal liver in sheep carried an overall mortality rate of 73% (42). For IUSCT in fetal sheep, trans-uterine ultrasound-guided injection has been shown to achieve a higher rate of engraftment when compared to an open delivery procedure at hysterotomy (70% vs. 20%) (32), despite in both cases cells were delivered intraperitoneally, commonly the route of delivery for fetal blood transfusion in clinical practice although less commonly performed than cordocentesis (47). In our study, we did not observe any miscarriage related to the transplantation procedure. Our miscarriage rate was similar to that we have previously reported for both intra-amniotic and intraperitoneal gene transfer to fetal sheep (9). In comparison, another study of xenotransplantation of human cord blood and fetal bone marrow MSCs into fetal sheep had an overall 37.5% fetal loss rate (33).
In our experimental setting, mesenchymal progenitors were derived in 100% of the animals. In keeping with previous studies in sheep (20) and other species (16), the AFMSCs were maintained in feeder-free cultures, had a doubling time of approximately 36–48 hours, and displayed mesenchymal stem cell markers (CD44, CD58, and CD166), while being negative for macrophages, hematopoietic, and endothelial markers (CD14, CD31, and CD45). When cultured in conditional medium (48) these cells could differentiate into adipogenic and osteogenic lineages. The potential of differentiation and characterization of all the surface markers did not change even after viral transduction. After being transduced, expanded undifferentiated cells were autotransplanted without sorting for two reasons. Firstly, we were pleased by the relatively high level of gene transfer that was achieved in the AFMSCs. Secondly we wished to avoid clonal selection of the cells and to observe what could happen with minimal manipulation. While we have previously shown that AFS cells can be efficiently transduced using adenoviral vectors (22), lentivirus vectors have the advantage of allowing sustained transgene expression (36). We have shown that lentivirus vectors are able to efficiently transduce sheep AFMSCs. This is particularly relevant when considering that various progenitors could be isolated from the amniotic fluid, and have demonstrated the possibility to differentiate into myocardium, lung epithelial cell, smooth muscle, and neuron cells (3,15,24). More recently we have also reported that ckit+/LIN– selected AF cells have hematopoietic potential both in vitro and in vivo (18). As a consequence, autologous AF cells could be engineered and used to engraft a fetus affected by a congenital disorder in order to provide for expression of the damaged/defective gene.
The level of injected cells in fetal tissues that we observed was admittedly low. However, for some congenital diseases, such as severe hemophilia, only 1% levels of protein expression are required to ameliorate the severe phenotype (50). Thus, for many congenital diseases this may be sufficient to improve the phenotype or even effect a cure (37).
The number of expanded transduced amniotic fluid cells increased as the length of time available for cell culture increased (Fig. 5A). Interestingly, however, there was no evidence of a dose–response effect, since the number of injected cells did not correlate with the number of organs containing transduced cells (Fig. 5B, C). Moreover, in some of the cases, the number of organs in which the GFP protein was detected was higher than the number of organs in which GFP DNA was detected. It is likely that cells were generating a chimerism and were not present in the entire organ. Since different samples of the same organs were analyzed for DNA and protein, it is possible that some samples did not contain positive cells. Injected cells appeared to migrate mainly to the liver, heart, adrenal gland, and umbilical cord of the fetus, and to extraembryonic tissues such as the amniotic membranes, placentomes, and amniotic fluid. Cells delivered to the peritoneal cavity are likely to migrate into the blood stream, from where they can travel to the placenta and membranes via the umbilical vein in the cord. This study was performed in normal fetuses in which there is no engraftment advantage for the injected cells. Different results may be obtained in animal models of disease where damaged organs may provide a niche for injected cells to engraft.
The question of whether autologous IUSCT is superior to allogeneic cells is as yet unresolved. In a previous study in sheep of fetal transplantation of MSC derived from fetal liver, the level of bone marrow engraftment of autologous MSCs achieved was not significantly different from that observed after allogeneic-derived MSCs (0.16% vs. 0.56%). However, engraftment in the fetal liver was higher (0.65% vs. 0.23%) after autologous transplantation (42). Similarly, we observed a good number of injected cells in the fetal liver, confirmed also by the appearance of rare cells coexpressing GFP and CK18 or AFP. Further studies need to be conducted to evaluate the possibility that AFMSCs may engraft in the liver. This could be particularly relevant since hepatocyte differentiation may lead to the treatment of congenital metabolic disorders (31,40). Our results could be improved by preconditioning the cells in hepatocyte differentiation media (41,52) and by injection in the umbilical vein (44). The latter in particular might preferentially target the liver when compared with the widespread tissue migration observed after peritoneal injection in this study.
In addition, the interval between injection and scheduled postmortem was relatively short. We cannot exclude that differentiation may occur mainly (or only) by cell fusion and further studies are needed, possibly in a small animal model, to further investigate this aspect. Moreover, the cytoplasmic marker (i.e., GFP) in an autologous model does not distinguish cell fusion from differentiation. Finally, even the expression of hepatocyte markers does not fully address the ability of these cells to generate functional hepatocytes.
In summary, this study demonstrates widespread and systemic migration after fetal injection of transduced autologous AFMSCs in the sheep. We successfully cultured sheep AFMSCs, and achieved a high level of lentivirus vector-mediated gene transfer. Longer term follow-up studies are needed to investigate whether cell engraftment is occurring. Our results, however, demonstrate that this approach may hold promise for targeted treatment of severe early onset genetic diseases that can be diagnosed in utero.
Footnotes
Acknowledgments
This work was supported by Chang Gung Memorial Hospital in Taiwan CMRPG370541, CMRP G370542, National Science Council of Taiwan NSC 97–2314-B-182A-057-MY2, and Newlife foundation for disabled children research grant in UK SG/08–09/10. S.B., M.C., and A.G. were supported by Città della Speranza, Vicenza, Italy. We thank Michael Boyd and Tom Bellamy, Royal Veterinary College, for their assistance.