
Abstract
Amniotic fluid has drawn increasing attention in the recent past as a cost-effective and accessible source of fetal stem cells. Amniotic fluid-derived mesenchymal stem cells (AFMSCs) that display high proliferation rate, large spectrum of differentiation potential, and immunosuppressive features are considered optimal candidates for allogeneic repair of mesenchymal damaged tissues. In this study, ovine AFMSCs (oAFMSCs) isolated from 3-month-old sheep fetuses were characterized for their proliferation rate, specific surface antigen and pluripotency marker expression, genomic stability, and mesenchymal lineage differentiation during their in vitro expansion (12 passages) and after nucleofection. The high proliferation rate of oAFMSCs gradually decreased during the first six subculture passages while the expression of surface molecules (CD29, CD58, CD166) and of pluripotency-associated markers (OCT4, TERT, NANOG, SOX2), the in vitro osteogenic differentiation potential, and a normal karyotype were maintained. Afterwards, oAFMSCs were nucleofected with a selectable plasmid coding for green fluorescent protein (GFP) using two different programs, U23 and C17, previously optimized for human mesenchymal stem cells. Transfection efficiencies were ~63% and ~37%, while cell recoveries were ~10% and ~22%, respectively. Nucleofected oAFMSCs expressing the GFP transgene conserved their pluripotency marker profile and retained a normal karyotype and the osteogenic differentiation ability. Seven single clones with a GFP expression ranging from 80% to 97% were then isolated and expanded over 1 month, thus providing stably transfected cells with long-term therapeutic potential. The in vivo behavior of GFP-labeled oAFMSCs was tested on a previously validated preclinical model of experimentally induced Achille's tendon defect. The allotransplanted oAFMSCs were able to survive within the host tissue for 1 month enhancing the early phase of tendon healing as indicated by morphological and biomechanical results. Altogether these data suggest that genetically modified oAFMSCs might represent a valuable tool for in vivo preclinical studies in a highly valid translational model.
Introduction
Amniotic fluid represents one of the mammalian gestational extraembryonic structures that are rich sources of fetal cells. Over the last decades, human amniotic cells have been efficiently collected and used for prenatal genetic testing of a wide variety of chromosomal and monogenic diseases. Recently, amniotic fluid has gained great excitement in the field of clinical applications for two main findings: (1) the isolation of a population of cells that have features of pluripotent stem cells (19, 36, 38) and (2) the evidence of cell-free fetal nucleic acids in larger quantities than in maternal plasma (7). While this latter discovery will help in providing new insights into fetal development and novel biomarkers for diagnosis (35), the finding of amniotic fluid-derived mesenchymal stem cells (AFMSCs) adds a novel promising source of engraftable stem cells for regenerative medicine therapies (16, 55). Several reports have proved the intermediate nature of fetal AFMSCs between adult mesenchymal stem cells (MSCs) and embryonic stem cells (ESCs), because of their sustained self-renewal capacity, ability to differentiate into cells belonging to the three embryonic germ layers, low immunogenicity, lack of teratoma formation upon in vivo transplantation, and absence of ethical concerns (53). According to an international agreement, human MSCs are defined by three minimal criteria: (1) adherence to plastic ability; (2) positive expression of specific surface antigens (CD29, CD44, CD73, CD90, CD105) and negative expression of hematopoietic/endothelial markers CD45, CD34, CD14, CD19, and human leukocyte antigen (HLA)-DR; and (3) in vitro multilineage differentiation potential (21). Regarding the immunophenotype, fetal human AFMSCs place themselves once again between adult MSCs and ESCs, since they are positive for surface markers characteristic of mesenchymal and/or neural stem cells and also express octamer binding transcription factor 4 (OCT4), NANOG, and stage-specific embryonic antigen-4 (SSEA-4), which are markers specific of ESCs (1). The fetal origin have allowed AFMSCs to be reprogrammed into induced pluripotent stem cells (iPSCs) with higher efficiency than adult MSCs (26, 44). Most importantly, the absence of host immunoreactivity after the local or systemic delivery of AFMSCs represents an invaluable advantage for their use in nonautologous transplants that are aimed at the repair of structural and connective tissues, including bone, cartilage, fat, tendon, and muscle (1). Indeed, the use of adult MSCs for tendon/ligament regeneration has been widely investigated in humans and animal models, but the identification of the appropriate source of MSCs should still be determined (5, 42, 70). In this context of therapeutic applications, fetal AFMSCs could be of great importance for all the above-indicated peculiar features.
Two requirements are considered essential to verify the positive outcome of preclinical stem cell-based transplantation therapies: (1) an animal model that is strictly comparable to humans in terms of morpho/anatomical structures and related pathologies and (2) an effective method to identify transplanted cells within the injured tissue.
Regarding the first necessity, the commonly used rat and rabbit models of tendinopathies are unsuitable to predict clinical outcome in human trials (71); among larger mammalian species, the sheep represents an excellent animal model to inquire about safety, reliability, and effectiveness of cell-based treatments aimed at the healing of bone, tendon, and musculoskeletal defects. In fact, domestic sheep presents great affinity with humans for size, physiological functions, anatomic features, and mesenchymal regenerative processes (50, 52).
Concerning the method to locate engrafted exogenous cells, the use of genetically modified cells that express harmless fluorescent protein markers, such as luciferase (luc) or green fluorescent protein (GFP), is a well-established system (13). Given its lack of interference with cell functions, its bright luminescence and its easy detection even in living cells, GFP is considered among the best markers for short- and long-term monitoring of GFP-labeled transplanted cells (2).
The delivery of exogenous marker genes into primary culture cells is usually very hard to be achieved (13, 27); for this reason, a safe and efficient transfection method is required to genetically engineer stem cells. Among the current ones, methods that are based on defective virus (such as adenovirus, lentivirus, and retrovirus) are by far the most efficient; unfortunately, viral transduction of stem cells is considered not appropriate for therapeutic purposes, due to safety issues including insertional mutagenesis (31), virus itself immunogenicity (66), toxicity, and exposure to biohazards. As nonviral and safer gene delivery systems, both liposome- and electroporation-based methods have been employed to introduce transgenes and small interfering RNAs (siRNAs) into stem cells of different origins (14, 37). Recently, nucleofection, an electroporation-derived technology, has allowed efficient and direct delivery of foreign genes into the cell nucleus by using a combination of cell line-specific solutions and exposure to defined values of electrical pulses (27). This technology has been successfully applied for transgene delivery into several primary cell lines (48), embryonic stem cells, and adult mesenchymal stem cells (43).
The principal aims of this study were (1) to expand previously reported data (49) regarding the establishment and characterization of AFMSCs isolated from sheep fetuses (oAFMSCs), (2) to test nucleofection to efficiently introduce a GFP-expressing plasmid into oAFMSCs both in transient culture and in long-term culture of isolated stable clones, and (3) to investigate the in vivo properties of GFP-labeled oAFMSCs by using a validated preclinical model (51) of experimentally induced tendinopathy in sheep.
Materials and Methods
All chemicals and media were from Sigma, unless otherwise specified.
Isolation and Culture of oAFMSCs
The oAFMSCs were isolated and cultured as previously described (49). Briefly, sheep fetuses were collected at a local abattoir by removing the whole pregnant uterus and bringing it at 30°C to the laboratory for further evaluation and processing. Only fetuses of 25–35 cm of length, at approximately 3 months of development, were used. Once opened the uterus wall, ~100 ml of amniotic fluid (AF) was recovered with a sterile 20-ml syringe on a 18-gauge needle. oAFMSCs were centrifuged at 800 × g for 20 min to remove cell debris, and the resulting pellets were seeded in 250-ml flasks in growth medium (GM) composed of minimum essential medium α-transformation (α-MEM) (Gibco) supplemented with 20% fetal calf serum (FCS) (Lonza), 1% ultraglutamine (Lonza), 1% penicillin/streptomycin (Lonza), and 5 ng/ml fibroblast growth factor (β-FGF), as described (4). The cells were incubated at 38.5°C in 5% CO2. After 7 days of culture, the medium was replaced with fresh medium and was subsequently replaced three times a week. At 80% confluence, the flasks were washed to remove dead cells and debris, the cells were harvested with 0.05% trypsin-EDTA for 5 min at 38.5°C and plated again at 3 ×103/cm2 for 12 consecutive expansion passages.
Mitotic Activity and Cell Proliferation
The mitotic activity of the cells was recorded using the bromodeoxyuridine (BrdU) assay. The cells were plated at a density of 3 × 103/cm2, and when they reached 50% confluence, they were incubated with BrdU labeling medium (10 μmol/l) (5-Bromo-2′-deoxy-uridine Labeling and Detection Kit I, Roche) for 30 min at 38.5°C with 5% CO2. Paraformaldehyde fixed cells were incubated with anti-BrdU antibody at 38.5°C for 30 min, then immunostained with anti-mouse Ig-fluorescein for 30 min and DAPI (1:5,000/PBS) for 5 min. All samples were analyzed using an Axioskop 2 Plus incident-light fluorescence microscope (Zeiss) equipped with a CCD camera (Axiovision Cam, Zeiss) with a resolution of 1,300 × 1,030 pixels, configured for fluorescence microscopy, and interfaced to a computer workstation, provided with an interactive and automatic image analyzer (Axiovision, Zeiss). Digital images were acquired at 400x magnification using standard filter setup for fluorescein isothiocyanate (FITC) or DAPI. At least, five different plates were examined for each sample.
The proliferative activity of oAFMSCs, recovered from three different amniotic fluid samples, was analyzed at passages 1, 6, and 12 by calculating the population doubling time (i.e., the time during which the cell population doubled). The doubling time was calculated with the following formula:

where TD represents the cell doubling time, t represents the duration of cell culture, amd N0 and Nt represent the cell number after inoculation and the cell number after culturing for t hours, respectively.
Flow Cytometry
oAFMSCs derived from three different fetuses at different phases of expansion (passages 1 and 12) were evaluated by flow cytometry for the surface molecules and for intracellular stem cell markers.
For surface protein, 5 × 105 cells were pelleted and resuspended in 100 μl washing buffer [phosphate-buffered saline (PBS), 0.1% sodium azide, and 0.5% bovine serum albumin (BSA)] containing a saturating concentration of surface antibody and incubated in the dark for 30 min at 4°C with the following primary antibodies: anti-CD14 and anti-CD58 (LifeSpan Bioscences), anti-CD29 (VMRD), anti-CD31 and anti-CD45 (AbD Serotec), anti-CD49f (Beckman Coulter), anti-CD117 and anti-sex-determining region Y box 2 (SOX2; Abcam), anti-CD166 (Ancell), anti-OCT4 (Becton Dickinson), anti-TERT (telomerase reverse transcriptase; Calbiochem), and anti-NANOG (Chemicon International).
For intracellular antigens, before incubation with primary antibody, cells were incubated in fluorescence-activated cell sorting (FACS) lysing solution (BD) at room temperature (RT) in the dark for 10 min and then in Perm 2 (BD) at RT in the dark for 10 min. Primary antibodies were labeled using the appropriate FITC Zenon Antibody Labeling Kit (Gibco), following the manufacturer's instructions. Cells were analyzed on a FACSCalibur flow cytometer (BD) using CellQuest™ software (BD). Flow cytometer settings were established using unstained cells. Cells were gated by forward scatter to eliminate debris. To eliminate the possible auto-fluorescence of cells, the contribution of unstained cells in the measurement channel was removed. Twenty thousand events were counted for each analysis. All antibodies were titrated under assay conditions, and optimal photomultiplier (PMT) gains were established for each channel. Data were analyzed using FlowJo™ software (TreeStar). Mean fluorescence intensity (MFI) ratio was calculated dividing the MFI of positive events by the MFI of negative events.
Immunocytochemistry
For immunocytochemistry, oAFMSCs were cultured on glass cover slips, fixed in 4% paraformaldehyde for 10 min at room temperature (RT), and then washed with 0.05% Tween 20 in PBS. Nonspecific binding was blocked with 1% BSA in PBS for 1 h at RT. Cells were incubated overnight at 4°C with the following primary antibodies: anti-SOX2 (1:200; Abcam), anti-TERT (1:250; Calbiochem), and anti-NANOG (1:1000; Millipore). After three washes, the secondary antibody was added [cyanine 3 (Cy3) or Alexa Fluor 488-conjugated anti-rabbit antibody 1:500] and incubated for 1 h at RT. Nuclei were counterstained with DAPI (1:5,000; Vectastain) for 10 min. Images were captured using the Axiovision Cam Zeiss.
RNA Isolation and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from oAFMSCs using TRI reagent following manufacturer's instructions. The RNA was treated with DNaseI digestion for 15 min at RT. One microgram of total RNA was used for reverse transcription reaction with Oligo dT primer and BioScript™ (Bioline). 2X Ready Mix™ Taq PCR reaction mix was used for PCR reaction using 3 μl of cDNA and 0.5 μM of each primer, in a final volume of 25 μl. The PCR primers and parameters are shown in Table 1. The reaction mixtures were incubated for 5 min at 95°C, followed by 95°C for 30 s, 55°C for 30 s, 72°C for 45 s, and 72°C for 7 min. The PCR products were separated on 2% agarose gel stained with ethidium bromide, visualized on a Gel Doc 2000 (Biorad), and analyzed with Quantity One 1-D Analysis software (Biorad). RT-PCR was normalized by the transcriptional levels of the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene. Each PCR reaction was carried out in triplicate.
PCR Primers Used for RT-PCR Analysis

Karyotype Analysis
Cytogenetic evaluation was performed in non-nucleofected oAFMSCs at passages 1, 6, and 12 and in nucleofected cells at passage 6. Confluent cells were harvested from flasks, transferred in amino dish, and then incubated at 37°C for 24–48 h in Chang medium (Irvine Scientific). Metaphase cells were arrested with 0.1 μg/ml colchicine for 2 h at 37°C. Then oAFMSCs were detached from dishes, resuspended in a hypotonic solution (0.075 M KCl), and incubated for 20 min at 37°C. Cells were centrifuged at 1,000 rpm for 10 min and fixed in methanol/ glacial acetic acid (3:1) for 10 min. Chromosome spreads were obtained by dropping the cell suspension onto ice-cold glass slides and air dried. Metaphase chromosomes were subjected to GTG banding (Giemsa staining). A median of 30–40 metaphases were examined for each sample at approximately the 350–450 band level using a Nikon Eclipse 80i microscope and analyzed by Genikon software (Nikon). A normal ovine karyotype consists in 27 pairs (2n = 54) of chromosomes. A chromosomal aberration was defined as clonal when at least two metaphases showed the same abnormality.
In Vitro Osteogenic Differentiation
To analyze the osteogenic differentiation property of oAFMSCs, cells at passages 1, 6, and 12 were cultured for 7, 14, or 21 days in growth medium (GM) or in differentiation medium (DM) represented by GM supplemented with 50 μM ascorbic acid, 10 mM β-glycerophosphate, 0.2 μM dexamethasone, and 10% FCS. Osteogenesis was assessed by evaluating Alizarin red staining, alkaline phosphatase (ALP) activity, calcein uptake, and the expression of two bone-related genes (osteocalcin and collagen type I) (Table 1). In brief, fixed oAFMSCs (cold 4% paraformaldehyde) were incubated in 40 mM Alizarin Red S for 30 min at RT before stopping the reaction with 80% acetone. ALP activity was processed and analyzed according to the manufacturer's instructions (BCP/NBT liquid substrate system). In brief, cells were fixed in 4% paraformaldehyde and incubated with phosphatase substrate reagent (2 mg/ml). The reaction was evaluated under an optical microscope. Calcein uptake was analyzed after a preincubation of oAFMSCs with calcein (1 μg/ml) for 48 h. The calcein incorporation in the matrix was qualitative and evaluated under a fluorescent microscopy.
oAFMSC Nucleofection
Nucleofection of oAFMSCs was performed according to the optimized protocols for human MSCs provided by the manufacturer (Amaxa Biosciences). Briefly, about 6 × 1 0 5 cells were resuspended in 100 μl of human MSC nucleofector solution and nucleofected with or without 2.5 μg of pAcGFP-N1 vector (Clontech). In some experiments, the linearized form of the same plasmid, obtained after an overnight (ON) digestion at 37°C with DrdI restriction enzyme (Biolabs), was used. The U23 (high transfection efficiency) and C17 (high cell survival) programs were tested on the Nucleofector II device. Immediately after, cells were plated onto 90-mm dishes, with growth medium. Twenty-four hours after nucleofection, the medium was changed to remove dead cells, and cells were analyzed for cell recovery and transfection efficiency. For the establishment of oAFMSCs that stably express GFP, neomycin treatment (400 μg/ml) was applied 48 h after nucleofection. The selection was carried out for further 2 weeks. Cell recovery, expressed as the percentage of number of viable transfected cells over the number of viable nontransfected cells (control), was determined by trypan blue exclusion method. Transfection efficiency was based on the percentage of cells expressing GFP assessed by flow cytometry. oAFMSCs were stained with propidium iodide (PI), resuspended in 500 μl of PBS, and then analyzed with the flow cytometer Coulter Epics XL (Beckman Coulter). The fluorescence of GFP was measured in channel FL1, while the fluorescence of PI was in channel FL2, both on a logarithmic scale. At least 10,000 events were acquired for each sample.
For clonal selection, after 4 weeks of culture under 400 μg/ml of neomycin selection, oAFMSCs were harvested by trypsinization. Two hundred microliters of cell suspension containing a total of 4 × 103 cells were used for serial dilutions in 96-well plates. At confluence, the wells with the highest percentage of fluorescent cells were chosen by fluorescence microscopy. Selected clones were plated in 12-well plates and, successively, into 90-mm dishes. Finally, GFP expression of each single clone was analyzed by flow cytometry, as described above.
Experimental Tendon Lesion, Transplantation, and Evaluation
In vivo experiments were carried out on a total of six 50-kg adult sheep of Appeninica breed according to the guidelines and approval by the Health Italian Ministery and by the Ethical Committee CEISA of the University of Chieti and Teramo. All surgical procedures were performed under general anesthesia. First, a bilateral core lesion was created in Achilles tendons under ultrasound (US) guidance by injecting 400 UI of Clostridium histolyticum type 1A collagenase (Sigma) diluted in 0.5 ml saline (17, 18). In detail, the enzyme was injected in the middle portion of the tendon (5 cm proximal to the calcaneal tuberosity). Twenty days later, the enzymatically induced tendon defects were identified with the aid of US: one limb was injected with a suspension of 2 × 106 oAFMSCs in 0.5 ml of saline solution while the contralateral with saline alone (control tendon, CTR). The experiments were performed in three animals for each group (cell-treated and control). After surgical procedures, the animals were maintained under daily observation for signs of discomfort. Thirty days later, the animals were euthanatized. The explanted tendons were randomly divided and used for the biomechanical tests or for the morphological/biochemical analysis.
For the biomechanical tests, three samples for each tendon group were considered. The tendons were harvested at the calcaneus and musculotendinous junction and firmly ripped by serrated grips 2 cm away from the edge of repair site and the remaining free length of all specimens. The maximum specific rupture force (N/mm2) and the maximum stiffness (expressed as % of preloaded sample) were evaluated using a digital dynamometer (Tinius Olsen H10KT dynamometer). Before measurements, specimens were preloaded for 1 min with 10 N to provide initial tension (11). The displacement rate was 6 mm/min. Specimens were kept moist throughout the test period with isotonic NaCl solution prewarmed at 38°C. The morphological analysis was performed on the isolated explanted tendons transversally cut at least 5 mm far from the injured area. The specimens were placed in liquid nitrogen. Cryosections, 7 μm in thickness, were processed for hematoxylin–eosin (H&E) and Herovici (HE; a polychrome stain for differentiating precollagen from collagen) in order to describe the recovery of tissue architecture. In parallel, immunohistochemistry studies were performed to analyze the process of extracellular matrix remodeling. To this aim, primary anti-collagen 1 (COL 1; 1:200 PBS/1% BSA; Chemicon Int.) and 3 (COL 3; 1:500 PBS/1% BSA; Chemicon Int.) antibodies were used. Both antigens were revealed with secondary anti-mouse Alexa Fluor 488 (diluted 1:400 PBS/1% BSA; Molecular Probes) antibodies, while section cell nuclei were counterstained with DAPI. Moreover, sections with retrieved GFP-labeled oAFMSCs were immunostained for COL 1, as described above, and the antigen was revealed with an anti-mouse Cy3-conjugated (diluted 1:400 PBS/1% BSA; Millipore) secondary antibody. Morphological sample examinations were performed with an Axioscop 2plus epifluorescence microscope (Zeiss) equipped with a cooled color charge coupled device camera (CCD; Axiovision Cam, Zeiss) interfaced with an interactive and automatic image analyzer (Axiovision, Zeiss) at 200x of magnification. Frozen sections were also used for mRNA extraction to detect the presence of the transgene mRNA in the host tissue by reverse transcriptase (RT)-PCR analysis using GFP-specific primers (Table 1).
Statistical Analysis
The normality of data was tested with Shapiro–Wilks test before proceeding with ANOVA test, followed when necessary by the post hoc Tukey's test (for parametrical data) or by Kruskal–Wallis test followed by Dunn's post hoc test (for nonparametrical data) (Microcal Origin 6.0 and GraphPad Prisms). The data were expressed as mean ± standard deviation (SD). A value of p < 0.05 was considered significant, and a value of p < 0.01 was considered highly significant.
Results
Characterization of oAFMSCs
The isolation and expansion of oAFMSCs was feasible using the plastic adherence ability of mesenchymal stem cells and selected culture medium. Cell adhesion occurred in 5–6 days after the harvesting. Two distinct cell types, differing in morphology and size, were initially evident: smaller cells showed a fibroblastic-like shape, while larger polyhedral cells displayed a typical epithelioid appearance (Fig. 1A). During the in vitro expansion, the fibroblast-like cells prevailed, thus resulting in a more homogeneous cell population (Fig. 1B). The proliferation rate of oAFMSCs gradually decreased during the first six passages dropping from ~75% (passage 1) to ~45% (passage 6) to then stabilizing on levels of ~40–50% at passage 12 (Fig. 1C). The population doubling time resulted to be comprised between 12 and 18 h, without increasing with passage number. Moreover, the doubling time was not affected by the plating efficiency that did not change with passage number. Finally, no structural aberration and a normal euploid ovine chromosomal complement of 54 (either XX or XY) were found in the majority of investigated cells at passages 1, 6, and 12 after Giemsa staining (Fig. 1D). Only in one male fetus, oAFMSCs at passage 12 showed the presence of four trysomic cell clones (55, XY, +22) out of the 40 cells analyzed (data not shown).

Characterization of ovine amniotic fluid-derived mesenchymal stem cells (oAFMSCs). (A, B) Phase-contrast microscopy (40x) image of oAFMSCs at passages 1 and 12, showing an heterogenous phenotype at passage 1 with larger epithelioid-like cells (indicated by arrows) (A) and a more homogeneous cell population after 12 passages (B). Scale bar: 25 μm. (C) Mitotic activity of oAFMSCs at passages 1, 6, and 12 calculated by bromodeoxyuridine (BrdU) labeling. During subculture passages, the proliferation rate of oAFMSCs gradually decreased from ~75% (passage 1) to ~45% (passage 6) and reaching levels of ~40–50% at passage 12. *p < 0.01 versus. P1. (D) Karyotype of oAFMSCs at passage 6, showing an euploid number of ovine chromosomes (54, XY).
The CD29 and CD58 surface antigens showed a high MFI ratio while CD166 a moderate expression at the beginning of culturing (passage 1) (Table 2). Then the MFI ratio of CD29 and CD58 decreased whereas the MFI ratio of CD166 remained stable. Conversely, oAFMSCs lacked expression of CD49f and of all the analyzed hematopoietic/endothelial markers (CD14, CD45, CD31, CD117) (Table 2).
Flow Cytometry Analysis on oAFMSCs

MFI ratio, the average of three different biological samples; ± standard deviation. Value of p is calculated versus passage 1 MFI ratio values. oAFMSCs, Ovine amniotic fluid-derived mesenchymal stem cells; MFI, mean fluorescent intensity.
The four markers NANOG, SOX2, OCT4, and TERT that are typical of pluripotent stem cells were evaluated at both mRNA and protein levels (Fig. 2). RT-PCR analysis showed a stable mRNA content for all four genes up to passage 12. In detail, high mRNA levels were evidenced for the TERT and NANOG genes, a moderate expression for SOX2, and a very low expression for OCT4 (Fig. 2D). Flow cytometry and immunocytochemical analyses provided more detailed information concerning both the content and cellular localization of the proteins encoded by these pluripotency-associated genes. Flow cytometry showed that freshly isolated oAFMSCs had a high MFI ratio for TERT and NANOG, a lower value for SOX2, and a very low MFI ratio for OCT4. After 12 passages, TERT, NANOG, and SOX2 increased their MFI ratio values, while the one of OCT4 further decreased (Table 2). Immunocytochemical analysis demonstrated a progressive translocation from the nucleus toward the cytoplasm of two of these proteins during the progression of in vitro expansion (Fig. 2A–C). In fact, while at the beginning of culture almost all the oAFMSCs displayed the proteins within the nucleus, after 12 passages TERT and SOX2 proteins moved toward the cytoplasm in a percentage that never overcame 30% (enlarged boxes in Fig. 2A, B).
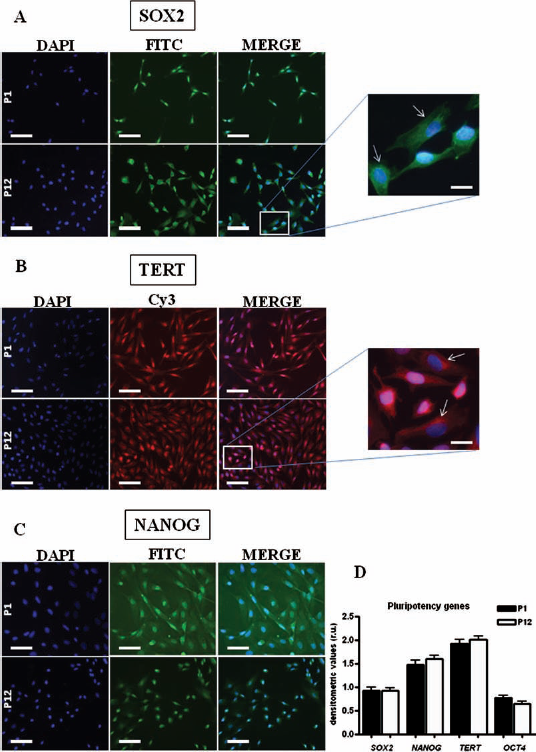
Expression of pluripotency-associated markers in oAFMSCs at the mRNA and protein level. (A–C) The immunocytochemical detection of sex-determining region Y box 2 (SOX2), telomerase reverse transcriptase (TERT), and NANOG proteins in oAFMSCs at passages 1 (P1) and 12 (P12) shows a progressive translocation from the nucleus toward the cytoplasm of SOX2 and TERT proteins during in vitro expansion (original magnification: 20x). Scale bar: 50 μm. In the enlarged boxes, the white arrows indicate the exclusive cytoplasmic localization of these proteins at passage 12 (original magnification: 100x). Scale bar: 10 μm. (D) The semiquantitative RT-PCR analysis of SOX2, NANOG, TERT, and octamer binding transcription factor 4 (OCT4) genes in cells at passages 1 (P1) and 12 (P12) showed a persistent and almost unchanged mRNA expression of all four genes up to passage 12. High mRNA levels were evidenced for TERT and NANOG, a moderate expression for SOX2, and a lower expression for OCT4. The expression levels of the pluripotency genes were normalized on that of the housekeeping glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene.
In Vitro Osteogenic Differentiation of oAFMSCs
To assess the in vitro mesenchymal differentiation potential of oAFMSCs, cells at passages 1 and 12 were incubated in osteogenic induction medium (DM) for 7, 14, and 21 days. A mineralized matrix started to be evident after 7 days as indicated by Alizarin Red and calcein uptake (Fig. 3A, C). The early in vitro osteogenic differentiation was also confirmed by ALP activity (Fig. 3B). Calcium deposition (Alizarin Red-positive nodular aggregates and calcein fluorescence) and ALP activity progressively increased as culture progressed (Fig. 3A–C). Neither or very low calcium mineralization or ALP activity was observed in control cells (CTR) cultured in growth medium (GM). The degree of matrix mineralization was also analyzed by evaluating the fluorescence of calcein (Fig. 3C). In osteoinduced cells, the amount of calcein uptake was higher than basal levels already after 2 weeks of culture in osteogenic medium and increased with progressive expansion. As expected, it was irrelevant in control cells that did not display any matrix formation. In all above-described analyses, there was no evidence of different behavior between cells induced to differentiate at passage 1 or passage 12 (data not shown).

Osteogenic differentiation of oAFMSCs at the protein and mRNA levels. (A–C) Osteogenic differentiation as determined by Alizarin Red S staining, alkaline phosphatase (ALP) activity, and calcein deposition at 20x magnification. Scale bar: 50 μm. A mineralized matrix (A, C) was evident after 7 days of culture in differentiation medium. The early osteogenic differentiation was also confirmed by ALP activity (B). Calcium deposition and ALP activity progressively increased as culture progressed up to 21 days. No calcium mineralization or ALP activity was observed in control cells (CTR) cultured in growth medium. (D–E) Expression profile of collagen type I (COL1A) (D) and osteocalcein (OCN) (E) genes. (D) The expression of COL1A mRNA in cells grown in normal growth medium (GM) after 7, 14, and 21 days were comparable with one of the cells analyzed at the beginning of differentiation (T0). The COL1A mRNA expression significantly increased in cells incubated for 7, 14, and 21 days in differentiation medium (DM) compared to the levels detected in cells expanded in normal GM and after 21 days reached the levels detected in adult bone tissue. *p < 0.01 versus, CTR. (E) The expression of OCN mRNA remained very low in cells grown in normal GM after 7, 14, and 21 days and similar to one of the cells analyzed at the beginning of culture (T0). The OCN mRNA expression significantly increased in cells incubated for 14 and 21 days in DM and after 21 days almost reached the levels detected in adult bone tissue. *p < 0.01 versus CTR.
The process of osteogenic differentiation was finally assessed at the mRNA level by evaluating the transcripts expression of two genes related to early and late phases of osteogenesis, such as collagen type I (COL1A1) (Fig. 3D) and osteocalcin (OCN) (Fig. 3E). The expression of both COL1A and OCN mRNA significantly increased in oAFMSCs incubated for at least 14 days in DM compared to the levels detected in cells expanded in normal GM, regardless of the passage number considered (data not shown).
Nucleofection of oAFMSCs and Generation of Stable Clones
Two different nucleofector programs, specifically designed for high transfection efficiency (U23 program) and high cell survival (C17 program) of human MSCs, were tested on oAFMSCs immediately after isolation (passage 0 or 1) to introduce the circular form of the selectable plasmid pAcGFP1-N1. Twenty-four hours upon gene transfer, oAFMSCs were examined for morphology, cell recovery, and transfection efficiency. No relevant morphological changes occurred in GFP-expressing oAFMSCs (Fig. 4A). GFP fluorescence signal was localized in both cytoplasmic and nuclear compartments with evident differences in intensity signals among cells, but regardless of the considered pulsing program. Trypan blue exclusion count indicated that nucleofection per se (carried out in absence of plasmid DNA) reduced cell recovery that dropped to 24.2 ± 1.1% and 49.7 ± 3.3% using the U23 and C17 programs, respectively. The percentage of viable cells further decreased in the presence of the plasmid, reaching 14.2 ± 6.1% (U23 program) and 22.3 ± 1.7% (C17 program). Flow cytometry analysis demonstrated that 62.7 ± 9.1% and 37.3 ± 2.5% of surviving cells exhibited GFP expression 24 h after nucleofection when programs U23 and C17 were used, respectively (Fig. 4B, C).

Characterization of nucleofected oAFMSCs as a pool (A–C, G) and as stable single clones (D–F, H). Morphology and green fluorescent protein (GFP) expression evaluated by fluorescent microscopy in nucleofected pooled cells (A) and in stable single clone D9 (D) at 20x magnification. The percentage of GFP-expressing cells, using the U23 program, as analyzed by flow cytometry in pooled cells was of 62.7% (B) and 96.6% (E) in single clone D9. The percentage of GFP-expressing cells, using the C17 program, as analyzed by flow cytometry in pooled cells was of 37.3% (C) and 92.6% (F) in single clone C8. Scale bar: 50 μm. Karyotype of nucleofected oAFMSCs as a pool (G) and single clone D9 (H) that revealed chromosomal stability after nucleofection and long-term culture. Results are representative of one independent experiment of both type of cells at passage 6.
In spite of the low cell recovery obtained with the U23 program, survived cells showed unaltered high proliferative potential (data not shown). The effect of plasmid vector shape on transfection efficiency was also evaluated by using the U23 program. GFP expression was ~2-fold lower when nucleofection was carried out with the linearized form of plasmid pAcGFP1-N1 (32% vs. 63%, linear vs. undigested circular plasmid, p < 0.01). Therefore, for the subsequent allotransplantation studies, the U23 program and the circular form of the plasmid vector were chosen.
In order to obtain a long-lasting expression of the GFP reporter gene, stable transfected clones were generated by limiting dilution method, under the selective pressure of antibiotic Geneticin. A few GFP-expressing oAFMSC clones were obtained for each nucleofection program used after two further weeks of antibiotic selection. Three and four stable clones, showing a diffuse and bright green fluorescence (Fig. 4D), were obtained for U23 and C17 programs, respectively, and clonally expanded in culture for more than 1 month. No stable transfected clones could be isolated from oAFMSCs nucleofected with the linearized DrdI-digested plasmid. Flow cytometry analysis revealed a maximum percentage of 96.6% and 92.6% GFP-expressing cells in the D9 (U23 program) and C8 (C17 program) clones (Fig. 4E, F) that were chosen for subsequent characterization. The percentages of GFP-expressing cells of the remaining five clones were 80.3%, 83.27%, and 93.1% for the C17 program and 80.8% and 95.5% for the U23 program (p > 0.05).
Assessment of Genomic Stability and Stem Cell Features of Nucleofected oAFMSCs
To evaluate whether nucleofection and long-term expansion of nucleofected cells could have affected oAFMSC phenotype and/or plasticity, GFP-expressing cells were analyzed for karyotype, pluripotency molecular markers, and in vitro differentiation potential. The structure and the modal number of chromosomes of nucleofected oAFMSCs were determined by karyotype analysis at passage 6 and after ~30 days of antibiotic selection. Pooled transfected oAFMSCs as well as each individual stable clone showed a modal chromosomal number of 54 (Fig. 4G, H), thus indicating the absence of gross chromosomal instability. The in vitro expansion of nucleofected oAFMSCs did not affect pluripotency marker expression, as shown by the protein and mRNA levels of SOX2, NANOG, and TERT (Fig. 5). Osteogenic differentiation, induced 2 weeks after antibiotic selection, indicated that nucleofection did not impair the differentiation features of oAFMSCs. In fact, the calcium deposition within the extracellular matrix (Alizarin Red S and calcein) (Fig. 6A, C), alkaline phosphatase activity (Fig. 6B), and the mRNA levels of the bone-specific COL1A and OCN genes (Fig. 6D, E) progressively increased when the cells were maintained under osteoinductive cultural conditions, similar to parental nontransfected oAFMSCs.

Expression of pluripotency-associated markers in nucleofected oAFMSCs as a pool. (A–L) Immunocytochemical detection of TERT, NANOG, and SOX2 proteins in nucleofected oAFMSCs at 20x magnification, showing no substantial differences with protein expression detected in parental nontransfected cells (already described in Fig. 3). Scale bar: 50 μm. (M) Semiquantitative RT-PCR analysis of SOX2, NANOG, TERT, and OCT4 genes in nucleofected oAFMSCs compared to oAFMSCs. The comparable expression at mRNA levels of SOX2, NANOG, and TERT genes between parental and nucleofected oAFMSCs shows that nucleofection and subsequent in vitro expansion did not affect pluripotency marker genes expression. The expression levels of the analyzed genes were normalized on that of the housekeeping GAPDH gene.

Osteogenic differentiation of nucleofected oAFMSCs at the protein and mRNA levels. (A–C) Osteogenic differentiation as determined by Alizarin Red S staining, alkaline phospatase (ALP) activity, and calcein deposition at 20x magnification. The calcium deposition (A, C) and ALP activity (B) increased with time when the nucleofected cells were cultured in osteoinductive medium. Scale bar: 50 μm. (D, E) Expression profile of collagen type I (COL1A) (D) and osteocalcein (OCN) (E) genes. The mRNA levels of the bone-specific COL1A and OCN genes progressively increased when the nucleofected cells were maintained under osteoinductive medium (DM) for 7, 14, and 21 days, compared to cells grown in standard medium (GM), and showed similar expression levels to the ones of parental nontransfected oAFMSCs. *p < 0.01 versus CTR.
Allotransplantation of GFP-Labeled oAFMSCs and Assessment of Functional Tendon Restoration
The allotransplantation of GFP-labeled cells was performed according previous studies that demonstrated the positive influence of transplanted and PHK26-labeled oAFMSCs on tendon healing (6, 51). To evaluate the behavior of GFP-labeled cells within the host tissue, the tendon core lesion were transplanted with 2 × 1 0 6 nucleofected cells (n = 3) or with saline solution (CTR) (n = 3). The morphological and functional tissue recovery was evaluated after 30 days. All animals well tolerated the surgical procedures. Analgesic pharmacological treatments were used during the first week after collagenase injection when an algic syntomatology was observed in all animals. By contrast, no inflammatory reaction was revealed immediately after cell allotransplantation or saline injection. All tendon explants treated with oAFMSCs did not show any inflammatory reaction while different degree of swelling characterized all CTR tendons. The morphological analysis performed with H&E and HE staining showed a more advanced level of tissue and matrix organization in oAFMSCs allotransplanted tendons (Fig. 7A–D). In detail, the repairing zone of CTR tendon showed several polyhedral cells that resulted dispersed amongst an abundant extracellular matrix where short new fibers were deposited with an irregular orientation (Fig. 7B, D). Conversely, in all tendons transplanted with oAFMSCs, the regenerated site displayed a microarchitecture that was similar to that observed in the healthy tissue with a more organized extracellular matrix, characterized by parallel fibers oriented along the longitudinal axis of tendon filled with fusiform aligned cells (Fig. 7A, C). The biochemical composition of extracellular matrix confirmed the stimulatory role played by GFP-labeled oAFMSCs. In fact, COL 3, the immature form of collagen, is normally secreted during the early phase of tendon healing and persisted irregularly distributed only in the repairing zone of CTR tissues (Fig. 7F). By contrast, in the oAFMSCs allotransplanted tendons, COL 3 was absent and the extracellular matrix resulted composed by aligned fibers of COL 1 (Fig. 7E, G). In detail, COL 1 was organized in fibers that, after 30 days, showed a typical orientation along the longitudinal axis of the tendon and with similar density of the healthy tissue. By contrast, in CTR tendons, this protein was weakly expressed, and it displayed a scattered random distribution throughout the repairing site (Fig. 7H).

Evaluation of allotransplantation of GFP-labeled oAFMSCs. Hematoxylin and eosin (H&E) and Herovici (HE) staining and immunohistochemistry (HI) for collagen proteins of injured Achille's tendon treated with GFP-labeled oAFMSCs or without cells (CTR). All images were photographed at 10x magnification, showing the healthy portion of the tendon under the white dotted line and the repair site within the upper portion. Scale bar: 100 μm. (A–D) H&E revealed an advanced status of regeneration in oAFMSC allotransplanted tendons with aligned fusiform shaped tenocyte-like cells with flattened nuclei entrapped within a regular extracellular matrix (A), composed as shown by HE staining of aligned collagen fibers, along the longitudinal axis of the tendon with a similar staining density to that recorded in the healthy tissue (C). By contrast, CTR tissue showed a disorganized structure with several polyhedral cells (B) dispersed in a scarce and irregularly distributed matrix within the repairing site (D). (E–H) The HI for the immature (COL 3) and mature (COL 1) matrix collagen proteins (green fluorescence) showed that in oAFMSC-allotransplanted tendons, COL 3 was barely detectable in the extracellular matrix, being replaced by COL 1 after 30 days (E, G). In fact, in the healing site COL 1 immunopositive fibers were abundant and parallel to the longitudinal axis of the tendon with a similar staining density of the healthy tissue (G). In CTR tendon, COL 3 immunoreaction revealed an irregular staining in the repairing site, and COL 3 fibers were not replaced by COL 1 fibers, since COL 1 was only weakly expressed (F, H). Nuclei were counterstained with DAPI. (I) Immunofluorescent microscopy (40x) image of GFP-labeled oAFMSCs that were recovered after 30 days from allotransplantation as green fluorescent polyhedral and fusiform cells entrapped within abundant COL 1 fibers (red fluorescence). One arrow indicates a GFP-labeled and fusiform cell that colocalized COL 1 protein (resulting in a yellow fluorescence). Two arrows indicate a polyhedral and immature GFP-labeled cell (green fluorescence). Nuclei were counterstained with DAPI. Scale bar: 25 μm. (J) RT-PCR analysis revealed the presence of GFP mRNA in expanded nucleofected oAFMSCs (lane 3) and in the healing portion of allotransplanted tendon (lane 1), while no expression was recorded in the CTR controlateral tendon (lane 2). M, molecular marker.
The long-lasting expression of the GFP transgene for 30 days at both protein and mRNA levels in the host tissue was also confirmed by immunohistochemistry (Fig. 7I) and by RT-PCR analyses (Fig. 7J). GFP-labeled oAFMSCs were always recovered within the engrafted tendon. The GFP signal indicated the presence of polyhedral and fusiform transplanted oAFMSCs that resulted entrapped among the new deposited extracellular matrix composed of COL 1. Some GFP-expressing cells, in particular those displaying a tenocyte-like fusiform shape, were able to colocalize the COL 1 protein (Fig. 7I, one arrow). No transgene mRNA expression was found in CTR tendons, following RT-PCR analysis (Fig. 7J, lane 2).
In parallel, the biomechanical test confirmed an acceleration of healing induced by the presence of transplanted oAFMSCs. In fact, the maximum failure load and stiffness that in damaged tendons reached values of 18 N/ mm2 (16–22 N/mm2) and 21% (15–28%), respectively, as a consequence of the induced defect, increased of 2.5 times (45 N/mm2, 41–49 N/mm2) and 1.7 times (35%, 32–40%) in the presence of cells and only of 1.6 times (29 N/mm2, 26–31 N/mm2) and 1.2 times (25%, 23–27%) in CTR tendons (p < 0.01). However, in 30 days the process of tendon regeneration resulted incomplete, since healthy tendons showed higher biomechanical performances [maximum failure load: 80 N/mm2 (74–88 N/mm2) and stiffness: 45% (39–50%)] (Fig. 8A, B).

Biomechanical tests. (A, B) Biomechanical properties (A, maximum failure load; B, stiffness) of CTR, damaged, oAFMSC-treated and healthy tendons. The maximum failure load and stiffness in damaged tendons were of 18 N/mm2 (16–22 N/mm2) and 21% (15–28%), respectively. In oAFMSC-treated tendons, both values increased 2.5 and 1.7 times, while in CTR tendons, they increased 1.6 and 1.2 times (p < 0.01). Healthy tendons showed higher biomechanical performances after 30 days posttransplantation. Data are expressed as minimum, maximum, and median values. Different superscripts denote statistically different values (p < 0.05).
Discussion
Due to their easy isolation, high proliferation rate, pluripotency features, and low immunogenicity, AFMSCs are a promising type of stem cells for allogeneic cell transplantation in different diseases of mesenchymal origin. Effective transplantation of adult MSCs for the repair of long bone defects (12, 22), craniofacial defects (40), and cartilage lesions (34) have already been reported in large animal models such as dog, sheep, and goat. Conversely, fetal mesenchymal amniocytes have been used in ovine models of diaphragmatic hernia (24, 67), lumbar spinal fusion (30), tracheal defects (41), and in rat model of bladder cryo-injury (20). Most recently, ovine autologous AFMSCs were transplanted in utero to show the feasibility of using these cells to treat congenital diseases before birth (65). Several studies have described the harvest and characterization of AFMSCs from different mammalian species, including human (19), mouse (5, 19), rat (20), pig (60, 64, 74), horse (46, 54), goat (32), rabbit (39), sheep (49), and buffalo (68).
The present study expanded the characterization of AFMSCs isolated from sheep fetuses and demonstrated the feasibility of the nucleofection technology to achieve short- and long-term GFP-labeling of oAFMSCs. Last but not least, the regenerative potential of these genetically modified cells was proved into a sheep flexor tendinitis model upon allogeneic transplantation.
The isolation of oAFMSCs by adherence property to plastic surface allowed to get significant amount of cells that could be cultured and expanded in vitro for at least 12 passages, without being altered in their phenotypical and functional features. At the beginning of their isolation, adherent cells were morphologically heterogeneous, consistent with human and mouse reports (19, 61), and with the nature of amniotic fluid that contains a variety of cell types deriving from skin, oral mucosa, airways, and digestive and urogenital tracts (56). With the increase of culture passages, a fibroblast-type morphology prevailed.
While the expression of a specific panel of surface antigens must be tested to phenotypically characterize human MSCs (21, 29), for other mammalian species the human antigens may not be suitable, and therefore a strict comparison between evolutionarily distant species is not always feasible. Conversely, an intraspecies immunological characterization of MSCs is much more reasonable. The surface marker profile of oAFMSC populations was in fact consistent with the described immunophenotype of ovine MSCs derived from bone marrow (oBM-MSCs) (50). At passage 1, oAFMSCs expressed the surface markers CD29, CD166 (similarly to oBM-MSCs), and CD58 while lacked the expression of cell membrane markers CD45, CD31 (similarly to oBM-MSCs), CD49, CD14, and CD117. Regarding the surface phenotype suggested for human MSCs (i.e., positive expression for CD105, CD73, CD90 markers and negative expression for CD45, CD14, CD34, CD19 markers) (21), the lack of cross reactivity of human antibodies with ovine cells and the lack of sheep-specific antibodies hampered an exhaustive comparison between these two species.
Undifferentiated and in vitro cultured oAFMSCs also expressed, for at least 12 passages, the pluripotent-associated molecular markers NANOG, SOX2, OCT4, and TERT, both at mRNA and protein levels. This result is consistent with similar studies carried out in porcine AFMSCs (74) and in buffalo AFMSCs up to passage 20, although this latter study was limited to only mRNA expression analysis (68).
Conventional karyotyping of oAFMSCs showed that most cells had a normal diploid chromosomal complement at passages 1, 6, and 12. Only in one fetus, four trysomic clones were detected at culture passage 12. Additional cytogenetic analyses on different independent cultures of the trysomic sample are needed to determine the nature of this second level mosaicism (i.e., cultural artifact or real mosaicism). In contrast to the widely reported genomic stability for long-term cultured human MSCs derived from bone marrow (BM) (59, 72) or amniotic fluid (19, 57, 59, 62), one study detected aberrant karyotypes at culture passages 11–14 in two of seven cases of human BM-MSCs (8). A detailed study on rat BM-MSCs showed a marked aneuploid karyotype even at early passages and a progressive chromosomal instability during in vitro expansion that were independent from cell culture conditions (23). Other two studies showed that the same typology of murine MSCs underwent to spontaneous transformation at passage 3 of in vitro culture, presenting evident chromosome instability both in number and in structure (25, 75). Since genomic instability has been associated to naturally occurring immortalization and cancer initiation, it is likely that rat MSCs cannot completely stand for human MSCs in clinical therapeutic approaches and that exhaustive molecular cytogenetic analyses are needed to investigate the actual potential of oAFMSCs.
The mesenchymal nature of oAFMSCs was proved testing their ability to differentiate into osteoblasts under in vitro specific culture conditions. The differentiation in osteogenic lineage was confirmed by standard functional analyses (Alizarin Red S staining, calcein uptake, ALP activity) and by evaluation of mRNA expression of the bone-specific genes OCN and COL1A.
In preclinical studies of tissue regeneration, the possibility of recognizing the implanted cells from endogenous ones represents a clear advantage. So far, only three methods (adenovirus, lentivirus, and liposome) have been tested to obtain genetically labeled AFMSCs with different outcomes. Viral transductions were able to yield stable cell clones with high efficiency and low cell mortality, using both adenovirus (28) and lentivirus vectors (65). To overcome the safety concerns associated to viral-mediated gene transfer, the chemical-based lipofection was used for the GFP-labeling of porcine AFMSCs to yield cloned embryos with higher efficiency than somatic cells (73). Among recently developed methodologies to transfer exogenous genes in primary MSCs, the nucleofection (27) and microporation (45) technologies are the most promising in terms of efficiency and cell surviving.
In this study, the nucleofection methodology was tested for the first time to introduce the GFP traceable marker gene in oAFMSCs. GFP-expressing cells were obtained with different transfection efficiencies and cell recovery rates, according to the nucleofector program used, and consistent with human MSCs results (3). Transgene expression was detected 24 h after nucleofection with an efficiency of about ~63% and ~37% using the U23 and C17 pulsing programs, respectively. In human BM-derived MSCs, comparable transgene expression levels were reached (i.e., ~74% using the U23 program and ~43% using the C17 one) (3). One disadvantage of nucleofection resulted to be the lower cell recovery in this particular type of mesenchymal stem cells, compared to the ones observed in hMSCs, namely ~10% versus 39% (U23 program) and 22% versus 44% (C17 program). Therefore, it might be necessary to optimize cell-specific buffer solutions and electrical pulse conditions for improving the recovery of viable nucleofected oAFMSCs. Nevertheless, the U23 program allowed to get viable cells that expressed high levels of GFP, continued to extensively proliferate, and could be used in subsequent preclinical transplantation applications. To this aim, it was of pivotal importance in the demonstration that they had maintained their genomic stability, pluripotency features and in vitro commitment towards osteogenic lineage, similarly to parental nontransfected cells. Furthermore, using both nucleofector programs and selective antibiotic pressure, seven stable transfected clones, expressing GFP for more than 1 month and without being altered in their features and differentiation abilities, were also obtained. In these clones, an almost complete GFP expression was detected with maximum expression levels ranging from ~80% to ~97%. However, transgene expression was not uniform in isolated clones, as previously observed (10). This finding is mainly due to the transgene random integration into the host cell genome and the so-called “chromosomal position effect” that can greatly affect the transcription levels of exogenous genes (15, 69). Since previous studies, carried out into fertilized mouse eggs, have shown that the transfection of linearized plasmids increases the efficiency of DNA integration and higher levels of stable functional integrants (9), a linearized plasmid was nucleofected into oAFMSCs to obtain the maximum percentage of living resistant clones that also express a functional GFP protein. However, in our experimental setting, halved transfection efficiency and no stable clone were obtained, suggesting that the linearized plasmid was mostly degraded by cytosolic nucleases. Further and more detailed experiments are needed to prove this hypothesis.
Overall, nucleofection technology was proved to be feasible to yield transient and stable delivery of a reporter gene in oAFMSCs, without impairing their features of pluripotent stem cells.
The in vivo regenerative potential of GFP-labeled oAFMSCs was then investigated upon their in situ allotransplantation into a preclinical model of induced tendonitis in adult sheep. While mouse models have several limitations (including short size and short lifespan) to be considered good animal models for preclinical studies, large animal models are more suitable given their morphological and functional similarities with humans. To this end, the sheep represents a valid alternative for preclinical development of therapeutic approaches aimed at the repair of elements belonging to the musculoskeletal system, including tendons. Since damaged tendons are unable of self-repairing, several studies have proved the utility of engrafting bone marrow- and adipose-derived MSCs for tendon regeneration (70), while the use of amniotic derived cells for tendon repair has only recently been proposed (6). Consistent with our previous evidences (6, 51), allotransplanted GFP-labeled oAFMSCs were able to engraft and survive into the injured ectopic sites for at least 1 month. In addition, the allotransplanted cells enhanced tissue regeneration and, most importantly, the recovery of mechanical properties during the early phase of healing. Further studies are needed to clarify whether the observed tissue ameliorations are due to stem cell ability to differentiate or rather to paracrine effects through the secretion of cytokines, adhesion molecules, proangiogenic factors, and other regulatory proteins (33, 47, 58). Since GFP-labeled oAFMSCs were shown to be well tolerated by the immune system of the host, this feature represents another important issue to be considered for the use of these cells in allo- and xeno-transplantation settings (51, 56, 63).
In conclusion, the reported data demonstrate that adherent cells isolated from ovine amniotic fluid and nucleofected with a GFP reporter gene, in a transient and stable prolonged manner, maintain the features of pluripotent stem cells and show a relevant regenerative ability for nonautologous tendon healing in large mammalian animal models.
Footnotes
Acknowledgments
This work was supported by Fondazione Tercas 2010. We want to thank Oriana Di Giacinto, Maura Turriani, and Francesco Mosca for their technical assistance. The authors declare no conflicts of interest.