
Abstract
Mesenchymal stromal cells (MSCs) are potent immunomodulators that have successfully been used to circumvent various types of inflammations, including steroid-resistant graft-versus-host disease. Although initially believed to be restricted to multipotent MSCs, this immunoregulatory function is shared with differentiated cells from the mesenchymal lineage such as skin fibroblasts (SFs). Mesenchymal cell-induced immunoregulation is so potent that it may allow the reactivation of dormant malignancies, a fact that would preclude using such cells as therapeutic agents. Because NK cells are pivotal effectors controlling tumor cell containment we investigated the effect of allogenic MSCs and SFs on NK cell function in vitro. When NK cells were incubated with IL-15 and MSCs or SFs for 6 days, their proliferation and cytotoxic activity were significantly decreased compared to NK cells cultured with IL-15 alone or with human venous endothelial cells. Cytotoxic activity inhibition reached 86% when assayed on MHC-I+ allogenic primary hematopoietic blasts, and was associated with a significant decrease in cytolytic granule exocytosis and in perforin release. Stromal cell-mediated inhibition was effective only if cell–cell proximity was long lasting: when NK cells were activated with IL-15 in the absence of MSCs and assayed for cytotoxicity in their presence no inhibition occurred. MSC inhibition was ultimately mediated by a soluble factor generated upon incubation with NK cells activated by IL-15 or IL-2. The indoleamine 2,3 dioxygenase was activated in MSCs and SFs because L-kynurenine was detected in inhibitory supernatants, but its blockade did not restore NK cell functions. The profound inhibition of cytotoxic activity directed against allogenic hematopoietic blasts exerted by MSCs and SFs on NK cells may be a concern. Should this occur in vivo it may induce the inability of NK cells to control residual or dormant malignant diseases after infusion of therapeutic MSCs.
Keywords
Introduction
NK cells are pivotal cellular effectors of innate immunity as illustrated by their prominent ability to eliminate virus-infected cells and to control tumor cell growth in healthy individuals. NK cells recognize their target via a combination of activating and inhibitory receptors encoded by germline DNA sequences (24). Once activated, NK cells secrete proinflammatory cytokines such as IFN-γ and TNF-α, and develop a potent cytotoxic function (5). This process comprises a sharp upregulation of CD107a [also known as lysosomal-associated membrane protein (LAMP)-1] expression on the cell membrane (1) followed by the exocytosis of preformed cytotoxic granules and the release of granule content on target cells. Perforin, a membrane-disrupting protein, and granzymes, a family of molecules structurally related with serine proteases, are released during this process. Perforin is the chief effector of the cytotoxic cascade as the effective delivery of the molecular cytotoxic mediators depends on its presence [for review see (43)]. Moreover, in humans, nonfunctional perforin is associated with a rare autosomal recessive disease called familial hemophagocytic lymphohistiocytosis (38).
The cytotoxic activity of NK cells can be exploited in clinical situations. For instance, upon the infusion of allogenic hematopoietic stem cells (HSC) in conditioned leukemic patients, passenger NK cells from the graft may help in eradicating the residual disease of the recipient in a process named ‘graft-versus-leukemia” (GVL) effect (29). However, NK cells also participate to the graft-versus-host disease (GVHD), which in some instances may be lethal, especially when it is refractory to chemical immunosuppressors. In such situations the infusion of mesenchymal stem cells (MSCs) represents the last resort to rescue patients (20,21).
MSCs reside in many human tissues (8), including the bone marrow where they provide hematopoietic stem cell survival and self-renewal signals (44). Under stress conditions, MSCs home to sites of injury whereby they decrease local inflammation and participate to tissue repair and regeneration (35). Therefore, MSCs may represent a potent tool for regenerative therapies (18,45). Moreover, they exert a powerful regulatory effect on allogenic and autologous T lymphocytes and NK cells (26,37,44). MSCs exert their antiproliferative effect via a MHC-independent process (2). Although initially believed to be restricted to multipotent MSCs, the immunoregultory function is shared by more differentiated cells from the mesenchymal lineage that have lost the ability to differentiate in several cell types in vitro (12,33). By contrast, stromal cells distinct from the mesenchymal lineage such as human venous endothelial cells (HUVECs) or neurons are not endowed, at least in vitro, with this property (14).
Resident MSCs also interplay with malignant cells. They block their proliferation and simultaneously provide a rescuing environment that may favor their subsequent evolution into overt metastases (19,23). Further information concerning the mechanisms and the amplitude of MSC-induced immunomodulation, and in particular its effect on the GVL, is required before considering using these cells for generalized therapeutic purposes.
In this study we investigated in vitro the effect of bone marrow MSCs and skin fibroblasts (SFs) on purified allogenic NK cells. We found that these cells prevented NK cell proliferation, IFN-γ production, and cytotoxic activity towards allogenic targets. The inhibition required a long-term coincubation of stromal cells with NK cells, as well as a close proximity, and resulted in the efficient inhibition of cytotoxic granule exocytosis and perforin release. The impact of mesenchymal cells on tumor growth is also discussed.
Materials and Methods
Reactants
Medium and Kits
Ficoll-Paque was from GE Healthcare Bio-Sciences AB (Uppsala, Sweden). NK cell isolation kit was from Miltenyi Biotec GmbH (Bergisch Gladbach, Germany). CD34+ cell selection system was from Dynal Biotech ASA (Oslo, Norway). 199 medium and IMDM were from Invitrogen Ltd (Paisley, UK). FIX-PERM kit was from BD Biosciences (San Diego, CA).
Antibodies
Unlabeled blocking anti-IFN-γ (clone NIB42) was from eBioscience (San Diego, CA). FITC-labeled: anti-CD45 (clone J33 mIgG1) was from Immunotech (Marseille, France); anti-CD107a (clone H4A3 mIgG1), anti-CD158a (clone HP-3EA mIgM), anti-CD158b (clone CH-L mIgG2b), and anti-granzyme B (clone GB11 mIgG1) were from BD Biosciences; anti-CD2 (clone S5.5 mIgG2a) was from Invitrogen Ltd; anti-NKG2D (clone 1D11 mIgG1) was from Serotec (Oxford, UK). RPE-labeled: anti-CD56 (clone AF12–7H3 mIgG1) and anti-NKp46 (clone 9E2 mIgG1) were from Miltenyi Biotech; anti-perforin (clone 5G9 mIgG2b) and anti-NKB1 (clone DX9 mIgG1) were from BD Biosciences; anti-NKG2A (clone Z199 mIgG2b) was from Beckman Coulter Inc. (Fullerton, CA). Biotin-labeled: anti-IFN-γ (clone B27) was from Serotec; anti-NKp30 (clone AF29–4D12 mIgG1) was from Miltenyi Biotech. Isotype controls: FITC- and PE-labeled mIgG1, mIgG2b, and mIgM-FITC were from Dako (Glostrup, Denmark). Biotin-labeled mIgG1 and mIgG2a-FITC were from Ancell (Bayport, MN). mIgG2b-PE was from BD Biosciences.
Cytokines and Chemicals
Thrombopoietin (TPO), FLT3-ligand (FLT3), stem cell factor (SCF), and IL-15 were from PeproTech EC Ltd. (London, UK). Endothelial cell growth supplement (ECGS), dithiothreitol (DTT), 7-amino-actinomycin D (7AAD), monensin, and brefelin A were from Sigma-Aldrich (Buchs, Switzerland), and the carbocyanine dye vybrant DiD, 5-carboxyfluorescein diacetate succinimidyl ester (CFSE) was from Invitrogen.
Cell Purification and Cultures
MSCs
Femoral heads that are routinely removed from patients undergoing total hip surgery were collected after informed consent of the donors. The use of these specimens for research purpose and the aims of the study were approved by the ethics committee of the Geneva University Hospital. MSCs were amplified as reported previously in IMDM medium complemented with 5% human platelet supernatant (CM) (10,40). After 3–4 weeks, the cell phenotype was analyzed by flow cytometry and MSCs were tested for multilineage differentiation (39) (not shown). Cells were used from passages 2 to 4.
Fibroblasts
Human foreskin fibroblasts, which are routinely used to amplify viruses, were a gift of Dr Kaiser, Laboratory of Virology, Geneva University Hospital. Fibroblasts from passages 2 to 4 were used in this study to avoid senescence that occurred generally after 10 passages in culture.
NK Cells
NK cells were isolated from buffy coats of healthy blood donors after Ficoll-Paque gradient and negative selection. NK cell purity was >90% as evaluated by FACS. Purified NK cells were cultured in CM containing 25 ng/ml IL-15. For coculture experiments, MSCs were seeded at 7 × 104/well in 24-well plates in 500 μl of medium and assays were initiated 48–72 h later, when MSCs had reached the value of 9 × 104/well. NK cells were then added at NK/MSC ratios of 1:1 and 4:1.
HUVECs
HUVECs were a gift from Dr Ferrari-Lacraz, Immunology Service, Geneva University Hospital. Cells were cultured in medium 199 supplemented with 20% FCS, 50 μg/ml endothelial cell growth factor, and 10 U/ml heparin. Cells were used from passages 2 to 4.
Hematopoietic Blasts
CD34+ cells were purified from umbilical cord blood by positive selection. CD34+ cells were cultured in IMDM medium supplemented with 10% FCS, DTT 100 μM, TPO 10 ng/ml, FLT3 25 ng/ml, and SCF 20 ng/ml for 1–2 weeks.
Flow Cytometry Analysis
Surface Molecules
Cells were processed as described previously (39). Analyses were run on a FACScan or FACScalibur analyzer from BD Biosciences, in PBS containing 2% FCS, 0.02% sodium azide, and 5 μg/ml of 7AAD to identify dead cells (31).
CD107a
CD107a-positive cells were detected according to Alter et al. (1). Briefly, NK cells were incubated with 15 μl/ml of CD107a-FITC mAb for 1 h at 37°C, before the addition of monensin at 6 μg/ml for 4 h at 37°C. NK cells were washed and stained with anti-CD56 PE-labeled mAb for 30 min at 4°C.
Intracellular IFN-γ
Brefeldin A (5 μg/ml) was added 4 h before the termination of the cultures. Cells were processed using the FIX-PERM kit, and labeled with biotin-labeled anti-IFN-γ plus streptavidin PE, and FITC-labeled anti-CD45.
Perforin and Granzyme B
Cells were stained with anti-CD56 biotin-labeled mAb plus strepavidin APC for 30 min at 4°C, fixed, permeabilized, and stained with PE-labeled anti-perforin, or FITC-labeled anti-granzyme B mAbs.
Proliferation Assay by CFSE Dilution
PMBC (4 × 108) were labeled with 500 nM of CFSE for 10 min at 37°C in PBS (22) and incubated overnight at 37°C. NK cells were then isolated as described above.
Nonradioactive Cytotoxic Assay and Cell Cycle Analysis
Cytotoxic activity was assessed according to Johann et al. (13) with slight modifications. Target cells were resuspended at 106 cells/ml in PBS 0.5% FCS and incubated for 10 min at 37°C with 0.5 μM of DiD, instead of D275 dye. NK cells and DiD-labeled cells were incubated for 4 h at effector/target (E:T) ratios of 1:1, 5:1, and 10:1. Cells were washed, stained with PE-labeled anti-CD56 mAb, and analyzed in the presence of 5 μg/ml of 7AAD instead of propidium iodide. Killed targets were defined as 7AAD+/DiD+/CD56- cells. Cell cycle analysis was done according to Garvy et al. (11).
L-Kynurenine Detection
Kynurenine was determined according to Kudo et al. (17). After precipitation and incubation with Ehrlich reagent the absorbance of the supernatant was read at 490 nm on a Vmax ELISA plate reader from Molecular Devices Corporation (Menlo Park, CA).
Statistics
Mann-Whitney nonparametric rank test was used when two series of data were compared. A value of p < 0.05 was considered as significant. For multiple comparisons, ANOVA with the Bonferroni correction was used after confirmation of the Gaussian distribution of the data using skewness and kurtosis fitting. Means ± SDs are shown.
Results
Allogenic MSCs and SFs Prevent NK Cell Proliferation Induced by IL-15
After 6 days of culture with MSCs in an initial ratio of 1:1 without any cytokine, 27 ± 18% of the NK cells were still alive whereas this value was 2.6 ± 1.6% in cultures without MSCs (p < 0.009, n = 6) (Fig. 1A, left panel). Accordingly, the frequency of hypodiploid apoptotic nonadherent NK cells was decreased in cultures containing MSCs (Fig. 1A, middle and right panels), suggesting that quiescent NK cells were rescued by MSCs.

Effect of MSCs, SFs, and HUVECs on NK cells. Ex vivo CFSE-labeled NK cells were cultured for 6 days with (white column) or without (black column) the above stroma in the presence or the absence of stimulatory cytokine. (A) Primary cultures in the absence of cytokines. Left panel: mean percentage of live cells after culture without or with MSCs. Mean of six experiments. ∗p < 0.009. Middle and right panels: one representative experiment out of three showing the percentage of hypodiploid apoptotic NK cells recovered after culture without and with MSCs. (B) Cell proliferation of NK cells after incubation for 6 days with IL-15, with or without MSCs (left panel), SFs (middle panel), and HUVECs (right panel) (n = 33, 6, and 3, respectively. ∗p < 10–4 for MSCs and ∗p < 0.009 for SFs. (C) DNA content of cells after culture in the presence of IL-15 without and with MSCs. Numbers are the percentages of hypodiploid cells and cells in the S-G2-M phase. (D) Frequency of IFN-γ-positive cells after 6 days with IL-15 and the addition of IL-12 (1 ng/ml) 20 h before the end of incubation; color code is as in (A). Mean of six and three experiments (∗p < 0.024) without and with MSCs, and of three experiments without and with SFs. SDs are shown.
When NK cells were stimulated with IL-15, the presence of MSC and skin fibroblasts (SF) prevented NK cell proliferation with a similar intensity, whereas the presence of HUVECs had no effect (Fig. 1B). On the mean, 83 ± 15% of the NK cells became CFSE low in the absence of MSCs, whereas this value dropped to 44 ± 23% in their presence (p < 10–4, n = 33) (Fig. 1B, left panel), thus corresponding to a 52% inhibition. Such a decrease in cell proliferation was in agreement with a decrease of the proportion of cells in the S-G2-M phases in the presence of MSCs (Fig. 1C). When SFs were present in the wells, the inhibition of NK cell proliferation was more effective than in the presence of MSCs and reached 68 ± 25% (n = 6, p < 0.009). Finally, both MSCs and SFs induced an extensive decrease of the frequency of IFN-γ-secreting NK cells (Fig. 1D).
NK Cells Activated in the Presence of MSCs or SFs Exhibit a Decreased Cytotoxic Activity
Cytotoxicity was assessed by FACS. CD56+ effector cells were excluded from the analysis and dead targets were identified as DID+ 7AAD+ cells (Fig. 2A). NK cells cultured in absence of stimulatory cytokines did not significantly lyse target cells (not shown). By contrast, after activation for 6 days with IL-15 or IL-2 NK cells lysed, at an E:T ratio of 1:1 MHC-I negative erythroleukemic K562 cell line with 45 ± 11% efficiency, and MHC-I+ primary cells such as hematopoietic blasts derived from CD34+ cord blood cells, and MSCs with a similar efficiency of 14 ± 7.3% and 14 ± 11%, respectively (mean ± SD of 10, 7, and 7 experiments) (Fig. 2B). When NK cells were activated with IL-15 in the presence of MSCs or SFs, their cytotoxic activity against K562 was decreased at all E:T ratios tested (Fig. 2C, left panel). By contrast, activation of NK cells in the presence of HUVECs did not alter their ability to lyse target K562 cells (Fig. 2C, right panel). MSC-induced inhibition of NK cell cytotoxic activity was also efficient when primary hematopoietic blasts derived from CD34+ cord blood cells were used as target, and reached 86% at an E:T ratio of 1:1 (Fig. 2D).

Cytotoxic activity of NK cells with or without conditioning with MSCs, SFs, or HUVECs. (A) Left panel: methodology. Cell suspensions recovered from the cytotoxic assay containing DID labeled target and NK cells were stained with anti-CD56-PE mAb, and plotted according to these two parameters. Single positive CD56bright cells (rectangular gate) were excluded from the subsequent analysis. Right panel: 7AAD fluorescence profiles of DID+ cells used as target (here K562 cells) were plotted. Targets incubated alone were used to establish spontaneous death frequency (open profile). 7AAD fluorescence of targets incubated with NK cells was plotted after gating out the CD56+/DID- effector NK cells (gray profile). (B) Cytotoxic activity of NK cells stimulated in the absence of any stroma with IL-15, tested against K562 cells (n = 10), hematopoietic blasts amplified in vitro from CD34+ cord blood cells (blasts, n = 7), and in vitro amplified allogenic MSCs (MSCs, n = 7). Horizontal bars are means. (C) Ex vivo NK cells were activated with IL-15 without or with MSCs, SFs, or HUVECs for 6 days at a ratio of 1:1. Cytotoxicity was assessed thereafter. Left panel: cytotoxic activity against K562 was tested at various E:T ratios after conditioning with MSCs or SFs, or none (no stroma) (one experiment representative of three). Right panels: mean values ± SD of cytotoxicity obtained either with (white columns) or without (black columns) conditioning of NK cells with MSC (n = 41, ∗p < 10–10), SF (n = 3), or HUVEC (n = 5) prior cytotoxic assay. (D) NK cells were preconditioned with MSCs or not and assayed for cytotoxicity using CD34+-derived hematopoietic blasts instead of K562 as target. Left panel: one representative experiment done at an E:T ratio of 5:1 and 1:1. Right panel: mean inhibition observed at E:T ratio of 1:1 (n = 6, ∗p < 0.009).
NK Cell Cytotoxicity Inhibition Induced by MSCs Requires Long-Term Coincubation and Is Reversible
We further investigated the effect of MSCs on NK cells by varying the time of preincubation of NK cells with MSCs prior to exposition to the target. When no preincubation with MSCs was provided (i.e., when NK cells activated with IL-15 in the absence of MSC were assayed for their cytotoxic activity on K562 in the presence of MSCs), no significant effect on the cytotoxicity was observed even when MSCs outnumbered NK cells (Fig. 3A). No inhibition occurred as well when NK cells were cocultured with MSCs in the presence of IL-15 for 4 days, and tested for cytotoxic activity immediately thereafter or after a further culture of 2 days with IL-15 in the absence of MSCs (Fig. 3B, left panel). When NK-MSC cocultures lasted 6 or 8 days the killing of K562 was inhibited if assessed immediately after incubation with MSCs. However, the cytotoxic activity of NK cells could readily be recovered if NK cells activated for 6 days in the continuous presence of MSCs were incubated for 2 more days with IL-15 alone prior to incubation with the target cells (Fig. 3B, right panel).
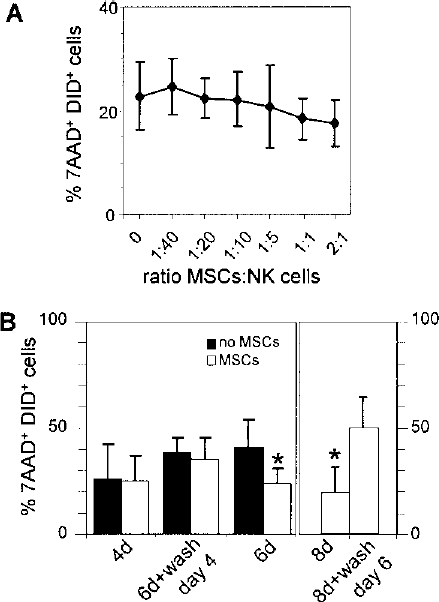
MSC-mediated inhibition of NK cell cytotoxic activity requires a prolonged coincubation. (A) Activated NK cells were incubated at a ratio of 1:1 with K562 cells and MSCs were immediately added to the coculture at MSC:NK ratios ranging from 1:40 to 2:1. The first 5 points are means of five, and the last 2 points of four independent experiments. SDs are shown. No significant difference according to Bonferroni multiple comparison statistics could be established. (B) Left panel: Purified NK cells were incubated with 25 ng/ml IL-15, with or without MSCs, for 4 days (4d) or 6 days (6d). NK cells from cocultures with MSCs were also collected after 4 days, and further stimulated with IL-15 in the absence of MSC for 2 days (6d + wash day 4). NK cell cytotoxicity towards K562 was assessed for the three different settings. Mean values of four independent experiments ± SD are shown, excepted for 6 days where n = 8 (∗p < 0.01). Right panel: activating cultures were also run for either 8 days with MSCs (8d) or 6 days with MSCs plus 2 days in their absence (8d + wash day 6), prior to assessing cytotoxicity on K562 cells. (∗p < 0.032, n = 5).
Both NK Cell-MSC Long-Term Proximity and Soluble Factors Are Required for the Establishment of Inhibition
Previous reports have suggested that an initial cell contact between MSCs and the target immunocompetent cells was required to induce the inhibition via a soluble factor synthesized by the MSCs (14,32). We investigated this issue by using transwells sealed with a 0.4 μm porosity membrane that prevented migration from one compartment to the other. MSCs were seeded in the lower chamber and NK cells in the insert. After 6 days of culture the inhibition of both NK cell proliferation and cytotoxicity was effective in the control conditions where cells remained in continuous proximity, but not in the transwell system (Fig. 4A). We then evaluated the inhibitory potential of supernatants harvested from primary cocultures with a documented inhibition (mean proliferation inhibition in primary cultures 30%, n = 6, p < 0.009). Once tested in secondary cultures on NK cells isolated from unrelated individuals, these supernatants efficiently inhibited NK cell proliferation. The inhibition produced by the conditioned supernatants in secondary cultures was more efficient compared to the inhibition observed in initial cultures (mean inhibition 54%, n = 6, p < 0.009) (Fig. 4B). The status of the IDO was assessed as this enzyme is known to be turned on in MSC activated by IFN-γ (16,32,34,36). Significant concentrations of L-kynurenine resulting from the degradation of L-tryptophan by IDO were detected in cultures of MSCs activated with IFN-γ and of MSCs incubated with NK cells stimulated with IL-15, indicating that IDO was functional. L-Kynurenine synthesis could significantly be inhibited with a blocking anti-IFN-γ mAb (Fig. 4C). This lead to a partial recovery of NK cell proliferation that did not reach statistical significance (Fig. 4D).

Cell contact and soluble factors are involved in the inhibition. (A) NK cells and MSCs were incubated for 6 days either as such in regular vessels (control) or in a transwell system with NK cells in transwells (transwell) and MSCs on the bottom. NK cells were tested for proliferation and cytotoxic activity against K562 cells. A significant inhibition of NK functions by MSCs is seen in control coculture conditions. ∗p < 10–4 and p < 0.003 for cell proliferation (% of CFSElow cells) and cytotoxicity (% of 7AAD+), respectively, but not in the transwell setting. n = 14 and 14, 8 and 6, for the determination of proliferation in control cultures, and cultures with 0.4 μm porosity membranes, and n = 9 and 9, 6 and 4 for the determination of cytotoxicity in control cultures, and cultures with 0.4 μm porosity membranes. Bars are SD. (B) Effect of conditioned supernatants. Supernatants from cultures that significantly prevented NK cell proliferation were tested in secondary cultures on freshly isolated NK cells. ∗p < 0.009 comparing proliferation with MSCs (white columns) or without MSCs (black columns); †p < 0.04 comparing the inhibitory effect observed in primary versus secondary cultures (n = 6). SD are shown. (C) Detection of kynurenine in various supernatants. Cross-ruled columns, MSCs cultured for 72 h without (MSCs) or with 500 U/ml IFN-γ (MSC-IFN-γ). Black column: NK cells alone stimulated with IL-15 (IL-15). White columns: NK cells cultured with MSCs, either without stimulation (NS), with IL-15 (IL-15), with IL-15 plus 10 μg/ml of IgG1 (IL-15 IgG1), and with IL-15 plus anti-IFN-γ mAb (IL-15 anti-IFN-γ) (∗p < 10−4, n = 9). Bars are SDs. (D) Proliferation observed in the same cell cultures as (C) supplemented with either control IgG or blocking anti-IFN-γ mAb. The difference observed is not significant (n = 8).
MSC-Induced Inhibition of NK Cell Cytotoxic Activity Is Associated with a Decrease of Lytic Granule Exocytosis and of Perforin Release
We investigated in more detail the impact of MSCs on the modulation of several molecules involved in the cytotoxic process. These included early mediators of cell-cell contact such as various NK cell receptors, intermediate effectors such as CD107a that is upregulated on the cell membrane after degranulation, and late effectors such as the enzymes perforin and granzyme B that are enclosed in the granules and participate to cytolysis once released in the cytolytic synapse.
After 6 days of activation with IL-15, with the exception of NKp46, the expression of most NK cell receptors was modulated compared to ex vivo values. NKB1, CD158a, and CD158b NKG2D and NKG2A were either down- or upregulated but with a similar amplitude irrespective of the presence or the absence of MSC. By contrast, NKp30 and CD2 exhibited a trend of upregulation in the absence of MSCs and a trend of downmodulation in their presence. However, due to the large dispersion of the values and the limited number of repeats these differences are not significant (Fig. 5A).

MSC impair cytolytic granule exocytosis and perforin release. Freshly purified NK cells were incubated with IL-15, with and without allogenic MSCs, at an initial ratio of 1:1 for 6 days. (A) NK receptor expression was analyzed by FACS and plotted as relative to ex vivo values normalized to 1. These data are the mean of two or three independent experiments. SDs are shown. (B) Membrane CD107a expression (left) and perforin content (right) prior to or after incubation with K562, with or without MSCs during preactivation. Black profiles: before incubation with K562; white profiles: after incubation with K562. (C) Mean values of similar conditions as (B). Black columns: activation of NK cells without MSCs; white columns: activation with MSCs; (–): before incubation with K562; (+): after incubation with K562; diff.: absolute value of the difference in either CD107a or perforin expression induced by incubation with K562 cells. These data are the mean of 14 and 10 independent experiments for CD107a, and 9 for perforin. ∗∗p < 10–3; ∗p < 0.02; and †p < 0.036 compared to the column immediately to the left. Bars are SD.
The median expression of CD107a and perforin was similar in NK cells cultured with IL-15 with or without MSCs. After incubation with K562 cells a significant increase of CD107a was observed in both types of cultures, but was significantly superior in cells activated in the absence of MSCs compared to cultures with MSCs (p < 0.041, n = 14 and 10) (Fig. 5B, C, left panels). Concerning perforin, its levels were significantly decreased after incubation with K562 cells in NK cells derived from cultures activated in the absence of MSCs, but perforin release was not significant in NK cells derived from cultures with MSCs (p < 0.041, n = 9) (Fig. 5B, C, right panels). Granzyme B was also released after incubation with target cells in NK cells activated in absence of MSCs, but due to a large dispersion of the data, no statistical significance could be established (not shown).
Discussion
In this report we investigated the effect of allogenic MSCs and SFs on NK cell activation and effector function with an emphasis on NK cell cytotoxic activity. Previous reports have investigated the impact of MSCs on purified NK cells (34,36,37) and of SFs on PBMC (14), but so far no report investigating the effect of human SFs on purified NK cells has been published.
When MSCs or SFs were incubated with NK cells for 6 days in the presence of IL-15, NK cells exhibited an altered cell proliferation and a decreased cytotoxic activity against a third-party target. These defects were specifically induced by MSCs and SFs, as HUVECs did not induce such alterations. Of note, IFN-γ-secreting NK cell frequency was decreased in the presence of MSCs, indicating that both NK cell effector function and their ability to stimulate bystander inflammatory cells were inhibited.
The inhibition induced by mesenchymal cells was reversible. Cell proliferation (not shown) and cytotoxicity were recovered if MSC-inhibited NK cells were removed from MSCs and further cultured with IL-15 for a couple of days (see Fig. 3). Moreover, MSC-induced inhibition was not immediate and required several days to be effective: no inhibition of NK cell cytotoxic activity occurred if activated NK cells were mixed with MSCs and target K562 cells simultaneously, or if NK cells were coincubated for 4 days only instead of 6 days prior to assaying their cytotoxic activity. Thus, these data show that the short-term contact of NK cells with MSCs does not provide immediate inhibition. Rather, as suggested by our experiments investigating inhibition kinetics or using conditioned supernatants from cocultures with previous documented inhibition, the synthesis of a soluble mediator that needs a rather long delay to reach functional levels is required for inhibition to occur. Nevertheless, our experiments with transwells indicate that cell–cell contact is required. It may well be that an initial cell–cell contact is required to induce the synthesis of the soluble molecule that will be the effector of NK cell inhibition. IDO was functional in our cultures, initially suggesting that L-typtophan starvation or L-kynurenine synthesis could be involved in the inhibitory process. However, IDO blockade, as confirmed by the significant decrease of L-kynurenine synthesis, did not lead to NK cell proliferation recovery. Attempts to inhibit prostaglandin synthesis with indomethacin and to block HLA-G or IL-10 with specific mAbs did not modify MSC-induced inhibition (not shown). We were thus unable to identify a single acting soluble inhibitor counteracting MSC effect. This is in agreement with previous observations suggesting that the inhibition exerted by MSCs on NK cells may be produced by the summation of the effects of several mediators (9,16,34,36).
One of our key observations was that cytolytic granule exocytosis, as monitored by CD107a expression, and perforin release were altered in NK cells activated by IL-15 in the presence of MSCs. Whether these alterations reflect the exhaustion of NK cell function provoked by MSCs is difficult to assess. The trend toward a decreased content in perforin observed in NK cells activated with IL-15 in the presence of MSCs prior to the cytotoxic assay compared to cells activated in control conditions (see Fig. 5C) supports the idea that NK cells consumed excessive amounts of perforin during their activation in presence of MSCs. Nevertheless, other mechanisms, such as MSC-induced inhibition of perforin synthesis, may also occur.
Of interest is that both CD107a expression upregulation and perforin release are mandatory for granule-mediated cytotoxicity to occur. If lytic granules do not fuse with the plasma membrane perforin cannot be released in the lytic synapse and cytotoxicity is impaired. Perforin is the master effector of granule-mediated cell death (6,38) and has no known molecular substitute. Indeed, defects in either lytic granule exocytosis or perforin release are associated with inheritable diseases in humans (6,38). Therefore, the inhibition induced by MSCs on these two processes represents a key impairment of NK cell effector function.
In the absence of stimulus MSCs rescued a significant proportion of NK cells from spontaneous death. These cells were quiescent, and readily proliferated upon subsequent activation in secondary cultures (not shown). Indeed, MSCs may enhance through this process the potential efficacy of the immune system by promoting the survival of immunocompetent quiescent progenitors (3,14,41), which may subsequently become fully functional if activated away from the MSCs. Unfortunately, MSCs also nurse malignant cells. Although they block the proliferation of various tumoral cell types, they simultaneously provide a rescuing, antiapoptotic environment (25). In the bone marrow MSCs impair the immediate development of malignant cells into sizable macroscopic tumors, but do not eradicate them. As for NK cells, this effect is reversible and malignant cells may remain dormant for many years, shielded from the immune system and cytotoxic drugs (23). Such oncogenic cells, often laden with an instable genome may, after an indefinite latency, develop into overt metastases (19). MSCs are also found in tumor-associated stroma whereby they deliver paracrine signals to tumoral cells that enhance their metastatic abilities (15). In this environment, their immunosuppressive effect may inhibit tumor-specific T or NK cells and allow tumor progression (4) or residual disease survival. Indeed, in this study we showed that the inhibition exerted by MSC upon NK cell cytotoxic activity was extremely potent, especially when tested on in vitro amplified hematopoietic blasts derived from CD34+ cells, where it reached 86%. Considering that allogenic hematopoietic blasts may mimic the residual disease that escaped from the patient conditioning, these observations suggest that in vivo infused MSCs, while inhibiting life-threatening GVHD, may significantly inhibit the GVL and participate to the relapse of the original malignant disease. However, if in vivo MSC-induced NK cell inhibition also vanishes once MSCs are no more present, as it is the case in vitro, one would expect that the GVL would recover once infused MSCs are diluted out in the organism.
MSCs aimed at a clinical application require, due to their low frequency in the initial cell suspensions, an extensive amplification in vitro. The cumulative number of population doublings generally exceeds 20 (42) and may lead to genome instability and eventual oncogenic transformation. Indeed, the literature is rather controversial concerning this topic with some authors describing a high rate of spontaneous transformation (27,28), while others, although observing occasional aneuploidy after amplification that seemed donor related, reported no transformation (30,42). Rather all cell lines tested in this study reached irreversible growth arrest after 35–52 population doublings. The reason of these discrepancies remain unidentified. Nevertheless, a recent survey performed on 227 patients infused by autologous MSCs amplified in vitro and followed up for 3–32 months could not detect any malignancies related to infused MSCs, suggesting that MSC infusion is safe (7). Moreover, several biological markers are now identified that allow to discard amplified MSCs that may be in the process of transformation before infusion to the patient.
MSCs may play a pivotal role in tumor progression and their therapeutic use should take into account the full spectrum of their biological activities. Our present work showed that bone marrow-derived MSCs very efficiently inhibited NK cell proliferation and cytotoxic activity. SFs were as effective as MSCs to induce such inhibitions, thus reviving older observations (12,33) and suggesting that fibroblasts, which are rather numerous in the organism, may represent an underrated cell population actively involved in immunomodulatory functions. Moreover, our work further suggests that we should keep on investigating the interactions of MSCs with immune and malignant cells to determine in which conditions they could be infused safely to patients.
Footnotes
Acknowledgments
This work was supported by grants from the Ligue Genevoise Contre Le Cancer (No. 0612) and from the Dr Henry Dubois-Ferriére Dinu Lipatti Foundation to V.K. and J.V. The authors thank Dr. J. Seebach and Dr. P. Walker for critically reviewing the manuscript, N. Brouwers and M. Barnet for excellent technical assistance, and Dr. D. Suva and Dr. P. Hoffmeyer for providing access to the femoral head samples.