
Abstract
MHC class I presentation of peptides derived from exogenous antigens (not synthesized within the antigen-presenting cell) is called cross-presentation and is mediated by selective subsets of dendritic cells (DC). A proportion of both donor and host DC may cross-present. Although there has been many studies that have investigated the role of donor versus host DC (i.e., direct vs. indirect pathway), what role cross-presenting DC play in allograft rejection has not been determined. We recently identified an agent, cytochrome c (cytc), that selectively depletes cross-presenting DC in vivo. By administering cytc we were able to study the impact of cross-presenting DC on rejection of islets grafted into fully mismatched mice. We found that cytc protected about half of the islet allografts from rejection. Our results indicate that cross-presenting DC can make potent contributions to the immune response to islet allografts, and contend that agents such as cytc that selectively target DC heralds a novel method of immunosuppression.
Introduction
Allograft survival is dependent upon continuous immunosuppression by a combination of drugs, such as tacrolimus and mycophenolate mofetil. This generalized immunosuppression predisposes the host to serious infections and cancer (19). In addition, these drugs have off-target (nonimmunological) effects. For example, rapamycin can suppress the bone marrow (3) and tacrolimus can be toxic to pancreatic islets and kidneys (13, 21,23,36). Allogeneic pancreatic islet transplantation can cure diabetes but an important limitation to its use has been these unwanted effects (37,41). The development of modalities that more specifically target the immune system and are less noxious would be a major advance.
Allograft rejection is critically dependent on dendritic cell (DC)–T cell interactions. Both donor and host DC contribute to allogeneic T-cell responses but play distinct roles (26). The conventional view is that donor DC (carried as passengers in the graft) present allogeneic MHC molecules directly to host T cells, resulting in potent T-cell responses and acute rejection. This is widely interpreted to reflect a high frequency of T cells specific for donor MHC antigens. Host DC have been ascribed a role in indirect presentation of graft-derived antigens that have been processed by the classical class II pathway (2) or by the class I cross-presentation pathway (8,18,28). More recent studies suggest that host DC also acquire intact donor MHC molecules for presentation to T cells, the so-called semidirect pathway (39). Indirect responses, while generally thought to be weaker than direct responses, play a central role in chronic graft rejection, and can promote the rapid rejection of grafts in some circumstances (2,17,42). Previous graft models that study the contribution of indirect responses to allograft rejection have relied upon physical or functional depletion of host or donor DC and/or CD4+ or CD8+ T-cell deficiency. In addition, indirect responses are frequently studied in the absence of major MHC antigenic differences. As such, these models do not always preserve the complex interactions between host and donor DC and T cells that exist in allotransplantation.
It should be noted that direct versus cross-presentation (terms often used in immunology) is not synonymous with direct and indirect pathway of presentation (terms often used in allo- and xenotransplantation), although there are some overlaps. Cross-priming/presentation was coined by Bevan (8) to describe when antigen from an exogenous source is processed to peptides presented on MHC class I molecules. Thus, both donor DC (direct pathway) and host DC (indirect pathway), or at least a proportion of them, may have cross-presenting ability, although the indirect pathway for stimulating host CD8+ T cells must only occur via cross-presentation. Therefore, the previous above studies that consider the direct and indirect pathway do not distinctly address the role of cross-presentation. In the current study, by selectively abrogating cross-presentation in vivo (see below), a more straightforward evaluation of the role of cross-presenting DC in allograft rejection has been availed.
Cross-presentation into the MHC class I pathway (presentation of exogenous antigens) has received much attention recently, as it explains how cytotoxic T-cell responses can be elicited when antigens are not endogenously expressed by professional antigen-presenting cells (e.g., viruses that do not infect such cells) (18,28). Exogenous proteins are taken up into the endosome by many cells including macrophages and DC, but only cross-presenting DC can transfer exogenous protein from the endosome to the cytosol (28). In the intrinsic pathway of apoptosis, cytochrome c (cytc), which normally resides in mitochondria, can only initiate apoptosome formation (by binding apaf-1 and procaspase 9) when it is released into the cytosol (22,33). Therefore, we reasoned that iatrogenically administered cytc would lead to the suicide of only cross-presenting DC. Indeed, we found that cytc injected into C57Bl/6 mice resulted in numerical reduction in the cross-presenting DC subset, and that this was dependent on apaf-1 (27). In the current study we have examined the potential of cytc to protect islet allografts from rejection.
Materials and Methods
Mice
C57Bl/6 (H-2b, abbreviated B6), CBA (H-2k), (B6×BALB/c)F1 (H-2b×H-2d), OT-I (20), OT-II (4), and CBA.RIP-Kb (40) mice were bred and maintained at the Walter and Eliza Hall Institute of Medical Research. All experimental procedures were approved by the Institution's Animal Ethics Committee.
Cytochrome c
Horse heart cytc (C2506, Sigma, St Louis, MO) was dissolved in saline at 20–50 mg/ml. All treatments were performed in vivo, and consisted of 5 mg doses of cytc administered according to the regimens described below.
Antibodies
Anti-CD11c (clone HL3), anti-CD4 (clone RM4-5), anti-CD8 (clone 53-6.7), and anti-TCRVα2 (clone B20.1) fluorochrome-conjugated monoclonal antibodies were obtained from BD Pharmingen (San Diego, CA).
Cell Lines
The pig kidney cell line PK15 (American Type Culture Collection) was stably transfected with DNA encoding a membrane form of ovalbumin (OVA) and designated PK15-OVA. Cells were maintained in 1 mg/ml G418 (Invitrogen, Carlsbad, CA) and 4 μg/ml puromycin (Sigma).
Flow Cytometry Analysis of DC Subsets
Mice were injected intravenously with a single dose of cytc or vehicle. After 21 h, spleens were harvested and digested for 25 min at room temperature with 1 mg/ml Type III collagenase (Worthington Biochemicals, Lakewood, NJ) supplemented with 0.14 mg/ml DNAse I Grade II (Roche Applied Science, Indianapolis, IN), then treated for 5 min with EDTA to disrupt cell complexes. Cells were stained with fluorochrome-conjugated monoclonal antibodies. DC were gated as CD11c+ cells. DC subsets, CD4+, CD8+, CD4–CD8–, were quantified by calibration against a known number of Calibrite beads (BD Biosciences, San Jose, CA).
In some experiments DC subsets were analyzed for their ability to phagocytose fluorescent microspheres in vivo. Mice were injected intravenously with vehicle or cytc. After 18 h mice were injected intravenously with 2.4 × 1010 Fluoresbrite YG carboxylate microspheres (0.5 μm; Polysciences Inc., Warrington, PA). Spleens were collected 3 h later and analyzed by flow cytometry as above.
In Vivo T-Cell Responses
OT-I (H-2Kb restricted OVA-specific) or OT-II (H-2I-Ab restricted OVA-specific) T cells were enriched (85–95% purity) from pooled lymph nodes of transgenic mice and labeled with the dye, carboxyfluorescein succinimidyl ester (CFSE, Invitrogen) (43). The T cells were injected intravenously into B6 mice such that each mouse received 1 × 106 OT-1 plus 1 × 106 OT-II cells. Two days later, antigen (PK15-OVA cells) was administered intravenously. Mice were either untreated or cytc treated intravenously 1 day prior and 1 day after antigen. Spleens were recovered 60 h after antigen injection, and flow cytometry was used to measure proliferation (CFSE dilution) in OT-I (Vα2+CD8+) and OT-II (Vα2+CD4+) T cells.
Islet Grafts
As diabetic hosts, we use CBA.RIP-Kb mice (40), which develop spontaneous, stable, nonimmune diabetes due to expression of a H-2Kb heavy chain transgene in their islet β-cells. Diabetic male CBA.RIP-Kb (H-2k) mice with a blood glucose level in excess of 20 mM were grafted with 400 fully allogeneic female (B6× BALB/c) F1 (H-2b×H-2d) islets under the kidney capsule. Islets were cultured for 2 days at 37°C and 10% CO2 prior to grafting. Mice with technically successful grafts (two blood glucose measurements less than 12 mM in the first week postgrafting) were subject to weekly blood glucose measurements. The time of graft rejection was defined as the first blood glucose measurement in excess of 12 mM with a subsequent return to pregraft levels in excess of 20 mM. Mice were treated with vehicle or cytc intravenously on the day prior to grafting then intraperitoneally daily for 2 or 7 days beginning on the day of grafting. At the end of the experiment, the graft plus kidney were fixed in Bouin's solution before sectioning and staining with hematoxylin & eosin and Gomori Aldehyde Fuchsin (GAF) for insulin granules.
Statistical Analyses
Statistical analyses were performed using Prism software (GraphPad Software Inc., San Diego, CA). Results of flow cytometric analyses are presented as mean + SD with comparison by one-tailed t-test. Graft survival curves were compared by a log-rank Mantel-Cox test.
Results
Cytc Preferentially Depletes CD8+ DC
We previously showed that cytc treatment depleted cross-presenting DC in B6 mice (27). As our islet allograft model uses spontaneously diabetic CBA mice as hosts, we wanted to test the effect of cytc treatment on DC in these mice (Fig. 1). DC (CD11c+) subsets were defined as CD8+, CD4+, or CD4–CD8– (Fig. 1a) and quantified by calibration against a known number of Calibrite beads (Fig. 1b–d). In vivo treatment of B6 mice with cytc resulted in a dramatic reduction in CD8+ DC (3.3-fold reduction, p < 0.001) (Fig. 1b) with a modest effect on CD4+ DC (1.7-fold reduction, p = 0.034) (Fig. 1c). Numbers of CD4–CD8– DC was not affected (Fig. 1d). These findings are consistent with previous studies defining CD8+ DC as the major cross-presenting DC subset with a secondary role for other DC subsets (5,10,11,28). In CBA mice, there was a reduced number of DC compared with B6 (Fig. 1) and the effect of cytc was less dramatic. Nevertheless, there was a 1.5-fold reduction in CD8+ DC (p = 0.007) (Fig. 1b), 1.2-fold reduction in CD4+ DC (p = 0.041) (Fig. 1c), and no significant reduction in CD4–CD8– DC numbers (Fig. 1d).
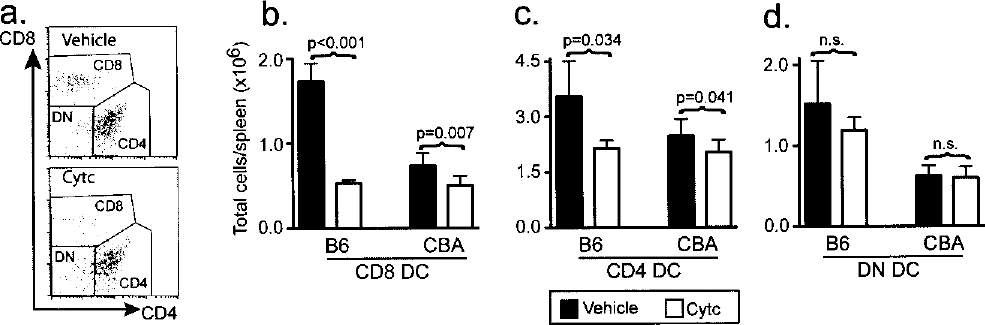
Flow cytometry analysis of spleen cells from mice treated with vehicle or cytc. (a) CD4+, CD8+, and CD4–CD8–subpopulations within the CD11c+ DC gate. The absolute number of (b) CD8+, (c) CD4+, and (d) CD4–CD8–DC per spleen CBA (n = 6) mice are shown as the mean ± SD and were compared by a one-tailed unpaired t-test. To confirm our previous findings, B6 mice (n = 3) were also investigated.
Cytc Preferentially Depletes Phagocytic DC
Phagocytosis by DC (although not sufficient by itself) promotes cross-presentation (16,34). Therefore, we wanted to examine the effect of cytc treatment on the phagocytic capacity of DC (Fig. 2). For this purpose, 18 h after the mice were treated with vehicle or cytc, fluorescent beads were injected intravenously and spleens were harvested 3 h later. DC that had phagocytosed beads could be readily identified by FACS (e.g., bead uptake by CD8+DC in CBA mice) (Fig. 2a). The percentage of bead positive DC was reduced by 2.3-fold in the CD8+ DC subpopulation (p < 0.001) (Fig. 2b) and 1.7-fold in the CD4+ DC subpopulation of CBA mice (p < 0.001) (Fig. 2c). A similar propensity for cytc to preferentially deplete phagocytic DC was also observed in B6 mice (data not shown).

Flow cytometry analysis showing bead uptake by DC of mice treated with cytc or vehicle. (a) Bead positive and bead negative subpopulations within the CD8+CD11c+ DC gate. The percentage of bead positive (b) CD8+CD11c+ DC and (c) CD4+CD11c+ DC in CBA mice (n = 6) are shown as the mean ± SD and were compared by a one-tailed unpaired t-test.
Cytc Selectively Inhibits CD8+ T-Cell Responses to Cross-Presented Antigen
We also wanted to confirm that cytc-mediated depletion of DC resulted in functional inhibition of CD8+ T-cell responses to cross-presented antigen while leaving CD4+ T-cell responses intact. As read-outs, we used T cells with a uniform T-cell receptor: OT-I (which sees OVA257–264 peptide on H-2Kb) and OT-II (which sees OVA329–337 peptide on IAb). A mixture of CFSE-labeled OT-I and OT-II T cells was injected into B6 mice. Antigenic challenge by injection of PK15-OVA transfectants resulted in OT-I and OT-II proliferation as measured by CFSE dilution (identified by CD8 and CD4 staining, respectively) (Fig. 3a, b). PK15-OVA cells are pig cell lines and therefore lack mouse MHC molecules and cannot present OVA peptide directly to OT-I or OT-II T cells. Thus, this model measures indirect responses of CD8+ OT-I T cells to antigen presented by the MHC class I cross-presentation pathway and CD4+ OT-II T cells to antigen presented by the classical class II pathway. The CD8+ OT-I T cell proliferative response to cross-presented antigen was profoundly inhibited by cytc treatment (Fig. 3a), resulting in a sevenfold reduction in the expansion of these cells (p = 0.001) (Fig. 3c). In support of the contention that cytc treatment preferentially inhibits cross-presentation to CD8+ T cells, the CD4+ OT-II T cell proliferative response was not significantly reduced (Fig. 3b, d).

Representative flow cytometry plots showing proliferation of CFSE-labeled (a) CD8+ OT-I and (b) CD4+ OT-II OVA-specific T cells in untreated or cytc-treated mice. Events collected for proliferated and unproliferated cells are enumerated in the left and right halves of the plots, respectively. The absolute number of (c) CD8+ OT-I and (d) CD4+ OT-II per million splenocytes is shown as the mean ± SD (n = 3 OVA-challenged mice per group; p-values are shown for a one-tailed t-test comparing untreated and cytc treated groups). A naive mouse was used as a negative control.
Cytc Enhances Islet Allograft Survival
Finally, we showed that cytc treatment enhanced islet allograft survival. Diabetic CBA.RIP-Kb mice (blood glucose >20 mM) were grafted under the kidney capsule with fully allogeneic islets resulting in a reduction in blood glucose to <12 mM. The time of graft rejection was defined as the first blood glucose measurement >12 mM with a subsequent return to pregraft levels >20 mM and is plotted in Figure 4a. Cytc treatment at the time of grafting (daily cytc for 3 or 8 days starting on the day prior to grafting) was able to reduce the potency of the alloresponse. Thus, grafts were observed to be fully functional beyond 100 days in 55% (6/11) of cytc-treated compared to 14% (1/7) of vehicle-treated mice. Normoglycemic mice after cytc treatment had the expected histological outcome viz. there were abundant GAF+ islets and minimal inflammatory infiltration (Fig. 4b, c). Typically, the histology from vehicle-treated diabetic mice did not have surviving islets, but scar tissue instead (Fig. 4d, e).

(a) Percentage graft survival shown for cytc-treated (solid line, n = 11) and vehicle-treated (broken line, n = 7) was compared by a log-rank Mantel-Cox test. Histology of (b) and (c) a surviving graft in a cytc-treated mouse and (d) and (e) a rejected graft in a vehicle treated mouse greater than 100 days postgraft. Hematoxylin & eosin stains are shown in (b) and (d). GAF stains are shown in (c) and (e); islet β-cells are stained violet-purple from the insulin granules. Arrows indicate areas of infiltrate. Scale bars: 200 μm.
Discussion
We have shown that cytc selectively depletes cross-presenting DC, thereby inhibiting CD8+ T-cell cross-priming while leaving CD8+ direct and CD4+ T-cell responses intact. Our previous study in B6 mice (27) was extended to CBA mice, our host of choice. CD8+ DC are the major resident cross-presenting population, although other DC subsets can cross-present albeit less efficiently (5,10,11,28). Consistent with this tenet, we found that CD8+ DC were most markedly depleted by cytc treatment in both B6 and CBA mice. As in our previous study, cytc depletion of DC, including CD8 DC, was incomplete, supporting the contention that phenotypic markers such as CD8 are on their own inadequate indicators of cross-presenting function. Thus, we confirmed that cytc-treated B6 mice lacked DC able to cross-present cell-associated OVA to OT-I CD8+ T cells while antigen presentation to OT-II CD4+ T cells was preserved [(27) and current study]. Furthermore, efficient phagocytosis has previously been identified as a functional marker of cross-presenting DC in a maturational state poised for efficient antigen uptake (43). In support of the functional relevance of cytc treatment in CBA mice we found that the phagocytic DC subpopulation was preferentially and markedly depleted.
We showed that depletion of cross-presenting DC provides long-term protection to a proportion of islet allografts. Because grafts depleted of donor leukocytes are frequently protected from allorejection, it has long been purported that indirect responses are relatively of minor importance (14,25,32). While CD8+ T cells clearly contribute to islet allograft rejection (12,38,44), failure to reject class I-deficient islets (29,30) has been used as further evidence of the minor role of the class I cross-presentation pathway. Furthermore, cross-primed CD8+ T cells are expanded on host MHC (whereas directly allo-primed CD8+ T cells are expanded on donor MHC) and therefore less likely to form cognate interactions with donor parenchyma bearing different MHC class I molecules. Thus, the prevailing wisdom is that cross-priming/cross-presenting DC are minor players in allograft rejection. Hence, it is perhaps surprising that cytc, by targeting cross-presenting DC, was able to protect allografts that differ from the host at MHC class I and II (as well as minor histocompatibility antigens). One would have expected that direct CD4+, indirect CD4+, and direct CD8+ T-cell responses should still occur and be sufficient to reject allografts.
There are several points to be considered about cytc-mediated protection from allograft rejection. Firstly, some grafts are rejected, which is what one may expect, if not all presentation pathways are blocked. Secondly, cross-presenting DC also partake in direct presentation so elimination of cross-presenting DC means there are also fewer DC to present directly. Thirdly, both donor and host cross-presenting DC may be affected by the cytc treatment. Migratory CD103+ DC with cross-presenting capacity have recently been identified in parenchymal tissues such as skin (6,9,15). Unfortunately, the numbers of donor DC within the islet graft are so meager that it is difficult to determine whether a partial depletion has occurred. Fourthly, the manipulations required to isolate the cross-presenting pathway in many previous models may result in an underestimation of the importance of cross-presenting DC, and indeed cross-primed CD8+ T cells can mediate rejection of allografts (17,42).
Our identification of cytc as an agent that selectively targets cross-presenting DC in vivo (27) uniquely enables reevaluation of the role of cross-presenting DC in a model that reproduces the most common clinical situation viz. grafting of tissues that are contaminated by donor passenger DC, differ at multiple major (both class I and class II) and minor histocompatibility antigens, and evoke CD4+ and CD8+ T-cell responses. These parameters have the potential to influence the allogeneic response to cross-presented antigen. DC migrating from inflamed tissues can act as a ferry to deliver antigen to cross-presenting DC in draining lymphoid tissues (1). By extrapolation, allograft studies using donor DC depletion to inhibit direct responses may unwittingly reduce antigen delivery to host DC and coincidentally reduce cross-priming. Antigen delivery to the cross-presenting pathway may also be reduced in models that rely upon grafting of MHC-deficient tissues in order to eliminate the direct response. Any allogeneic MHC-derived peptides that are epitopes for cross-primed CD8+ T cells will also be eliminated by the use of MHC-deficient grafts. Furthermore, graft damage mediated by the direct pathway may be an important contributor to the antigen supply to cross-presenting cells. In support of this, it has been shown that adoptive transfer of activated CD8+ T cells specific for ovalbumin antigen expressed in islets lead to increased cross-presentation in draining lymph nodes (24,31). Finally, efficient responses by cross-primed T cells may be dependent upon CD4+ T cells that license cross-presenting DC (7,35), such that the potential of cross-primed CD8+ T cells to reject allografts can only be fully assessed under circumstances where CD4+ T cells are available. Such complex interplay between antigen release, the different cell types, direct and cross-presentation etc would indicate that the elimination of antigen-presenting cells that mediate one pathway may have a greater effect than initially anticipated.
We conclude that cross-presenting DC play an important role in initiating allograft rejection. Moreover, the therapeutic use of cytc is amenable to other species including humans. Thus, the selective action of cytc on DC heralds a new pathway of immunosuppression.
Footnotes
Acknowledgments
This work was supported by National Health and Medical Research Council of Australia (NH&MRC), Diabetes Australia, Juvenile Diabetes Research Foundation, NH&MRC Independent Research Institutes Infrastructure Support Scheme grant #361646, and Victorian State Government Operational Infrastructure Support grant. We are grateful to Nicole Ashman for technical assistance with mice, and to Steven Mihajlovic for help with histology.