
Abstract
Islet transplantation can cure type 1 diabetes but is limited by lack of donor organs and early graft dysfunction, such that many patients require multiple transplants to achieve insulin independence. Monocyte-derived dendritic cells (moDCs) arise during inflammation and allograft encounters where they can promote various innate and adaptive immune responses. To determine whether moDCs impair early graft function following allogeneic islet transplantation, we transplanted MHC-mismatched BALB/c (H-2d) islets into diabetic C57BL/6-CCR2. DTR recipients (H-2b) treated with either saline (control) or diphtheria toxin (DT) to deplete moDCs. Graft function was assessed by blood glucose (BG) measurement. DT treatment resulted in specific depletion of graft site moDCs posttransplant. Despite equivalent pretransplant BG levels [27.0 ± 1.3 vs. 29.6 ± 1.1 mM, not significant (ns)], DT recipients achieved lower posttransplant BG levels and better rates of normoglycemia than control recipients (11.0 ± 1.9 vs. 19.1 ± 1.4 mM, p = 0.004) at 1 day posttransplant in diabetic recipients. When a suboptimal donor dose of 200 islets was transplanted, DT-induced moDC depletion resulted in normoglycemia in 78% compared to 25% of control recipients (p = 0.03). As well as amelioration of graft dysfunction in the immediate peritransplant period, prolonged DT administration (15 days posttransplant) resulted in improved graft survival (21 vs. 11 days, p = 0.005). moDCs impair early graft function post-allogeneic islet transplantation. moDC depletion may allow for improved early graft function, permit transplantation with lower islet masses, and enhance graft survival.
Introduction
Type 1 diabetes is a chronic autoimmune condition characterized by progressive destruction of the insulin-producing β-cells of pancreatic islets 1 . While insulin therapy mitigates many of the most severe acute complications, it imposes substantial lifestyle burdens, and significant risks of long-term complications such as retinopathy, neuropathy, nephropathy, and cardiopathy remain 2 . Islet transplantation can cure type 1 diabetes, leading to improvements in quality of life, avoidance of acute hypoglycemia, and reduced risk of long-term complications 3 . Despite these benefits, islet transplantation has yet to become a routine therapeutic option due to a lack of donor organs, poor early graft function, and the adverse effects of long-term immunosuppression; thus, it remains reserved for a small subset of patients with hypoglycemic unawareness or poor quality of life 4 .
One of the major ongoing problems in islet transplantation is early graft dysfunction resulting from the significant stresses to which transplanted islets are subjected in the first few days posttransplant, prior to completion of the revascularization process 3 . Up to 60% of the graft may be damaged or lost during this period 5 . It is therefore not surprising that most patients who become insulin independent require more than one islet transplant 6 . Various factors have been found to adversely impact early posttransplant graft function, including the instant blood-mediated inflammatory reaction (IBMIR) 3 ; inflammatory mediators including interferon-γ (IFN-γ), interleukin-1β (IL-1β), or inducible nitric oxide synthase (iNOS)7,8; and inflammatory cells9,10.
Monocyte-derived dendritic cells (moDCs) have been recently found to increase in response to alloantigen exposure during experimental heart and kidney transplantation 11 . This occurs independently of tissue damage-associated signals 12 . To determine the role of moDCs in vivo, we had previously used CCR2.DTR mice, which express a diphtheria toxin receptor (DTR) under the control of the CCR2 promoter, therefore allowing conditional depletion of CCR2-expressing moDCs by DT administration13,14. Given the above developments, it was timely to use these mice to examine the effects of moDCs on graft function following allogeneic islet transplantation.
Materials and Methods
Mice
B6.50-1 15 , CCR2.DTR 16 , and BALB/c mice were bred under specific pathogen-free conditions in the animal facility at the Walter and Eliza Hall Institute. Experiments were performed according to the guidelines of the Institute's Animal Ethics Committee.
Induction of Diabetes
Diabetes was induced in recipient mice in two ways. First, streptozotocin (STZ; Sigma-Aldrich, St. Louis, MO, USA) was administered intraperitoneally (IP) at 250 mg/kg 4 days prior to transplantation. Alternatively, B6.50-1 mice develop spontaneous, stable, nonimmune diabetes due to expression of an H-2Kb heavy chain transgene in their islet β-cells. Diabetic mice were selected based on blood glucose (BG) readings >18 mM on at least two separate occasions.
Islet Isolation and Transplantation
The islet isolation and transplantation procedure has been previously described 15 . Briefly, pancreata were collagenase P digested (Roche, Indianapolis, IN, USA), and islets were enriched on Histopaque-1077 (Sigma-Aldrich) density gradients. Islets were handpicked and counted immediately prior to transplantation under the kidney capsule of anesthetized mice.
Assessment of Graft Function
Graft function was assessed by measurement of BG levels by tail vein cannulation using Accu-chek glucometer strips (Roche, Basel, Switzerland). BG levels were measured two to three times per week at predefined time points. Functioning grafts were identified by achievement of normoglycemia (BG <12 mM) after transplantation 17 . Graft failure was determined by return to BG >18 mM posttransplant on two consecutive occasions.
moDC Depletion
CCR2+ moDCs were depleted by IP administration of 20 ng/g DT at 2- to 3-day intervals for various durations from 3 days pretransplant up to 15 days posttransplant.
Dissociation of Islet Allografts
Mice were euthanized, and grafts were dissected away from the kidney prior to digestion at room temperature in 1 mg/ml type III collagenase (Worthington, Lakewood, NJ, USA) and 0.01% (w/v) grade II bovine pancreatic DNAse I (Roche) for 30 min with addition of ethylenediaminetetraacetic acid (EDTA) at a final concentration of 8 mM for the final 5 min.
Flow Cytometry
After IgG Fc receptor (FcR) blockade by 2.4G2 Ab (produced in-house), single-cell suspensions were stained with Ab-specific stain for CD11b (M1/70), CD11c (HL3), Ly6C (AL-21), major histocompatibility complex (MHC)-II I-A/I-E (M5/114.15.2), CD45 (30-F11), and Ly6G (1A8) from BD Biosciences (San Jose, CA), or F4/80 (BM8) from eBioscience (San Diego, CA). Analysis was performed on a BD FACSVerse. Data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Statistical Analysis
Mean and standard deviation (SD) values were calculated, and two-tailed Student's t-tests, chi-square tests, or log-rank (Mantel–Cox) survival analyses were performed with GraphPad Prism Software (La Jolla, CA, USA).
Results
Administration of DT to CCR2.DTR Recipients of Islet Allografts Results in Depletion of Graft Site moDCs
We defined moDCs according to surface phenotype as has been previously described: CD11b+CD11c+Ly6C+ MHC-II+(13,14). Furthermore, we applied CD45 as an additional marker to distinguish infiltrating leukocytes from graft and kidney parenchymal cells (Fig. 1A). We have previously found that DT administration to CCR2.DTR mice selectively depletes moDCs in lymphoid tissues13,14. To determine whether this treatment can also ablate moDCs at the graft site, we transplanted 400 allogeneic BALB/c islets into CCR2.DTR recipients that had been treated with either DT or normal saline 3 days and 1 day earlier. Grafts were recovered 1 day posttransplant and analyzed by flow cytometry. While total CD45+ and CD11b+ cell numbers at the graft site were not significantly different between the two groups, DT recipients had almost total ablation of moDCs compared to control recipients (Fig. 1B). To ensure that this result was not due to a nonspecific DT effect, we conducted the same experiment using CCR2.DTR and CCR2 wild-type (WT) recipients that were all treated with DT. Consistent with the results from the previous experiments, we observed a significant depletion of moDCs in the CCR2.DTR recipients compared to the WT controls at 1 day posttransplant (Fig. 2). We found no evidence of any differences in the numbers of CD4+ T cells between the two groups, and there were no CD8+ T cells detected in either group at this time point. Of note, while we did observe a reduction in monocyte numbers in DT-treated CCR2.DTR recipients (Figs. 1B and 2), this depletion was more variable and less robust. Hence, we concluded that the CCR2.DTR model could be used to study the effect of moDCs on islet transplants.

Administration of DT to CCR2.DTR recipients of allogeneic BALB/c islets results in effective depletion of moDCs from the graft site. (A) Gating strategy to identify and enumerate moDCs at the graft site. (B) Enumeration of viable CD45+ cells, CD11b+, moDCs, and monocytes from the graft site of CCR2.DTR recipients 1 day after transplantation of 400 allogeneic BALB/c islets with or without administration of DT. Flow cytometry plots in (A) are gated on total graft site cells. ∗∗∗p < 0.0001, ∗p < 0.05, two-tailed Student's t-test. Error bars indicate SD. Results are pooled from two independent experiments. DT, diphtheria toxin; CCR2.DTR, transgenic for the DT receptor under the CCR2 promoter; moDCs, monocyte-derived dendritic cells.
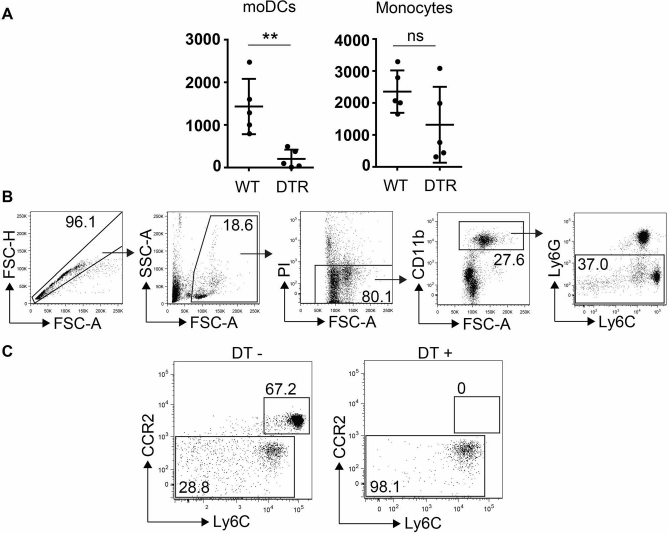
(A) Enumeration of moDCs, monocytes, and CD4+ T cells from the graft site of CCR2.DTR or CCR2.WT recipients 1 day after transplantation of 400 allogeneic BALB/c islets following administration of DT. (B) Gating strategy to identify monocyte populations in the peripheral blood of CCR2.DTR mice. (C) Effect of DT administration on CCR2+ monocyte and CCR2− monocyte populations in the peripheral blood of CCR2.DTR mice. ∗p < 0.05, ∗∗p < 0.01. DT, diphtheria toxin; CCR2.DTR, transgenic for the DT receptor under the CCR2 promoter; WT, wild-type; moDCs, monocyte-derived dendritic cells.
Depletion of moDCs Results in Improved Early Graft Function After Allogeneic Islet Transplantation in STZ-Induced Diabetes
To determine whether moDCs impair early graft function following allogeneic islet transplantation, we first induced diabetes in CCR2.DTR recipients by IP injection of STZ 250 mg/kg 4 days before transplantation. We treated these recipients with either DT 20 ng/g IP to deplete moDCs or normal saline as untreated control on day −3 or day −1 pretransplant. One day before transplantation and on the day of transplantation, we measured BG levels on all STZ-treated mice and used only mice with established diabetes (BG >18 mM on both occasions) to proceed to transplantation. Pretransplant BG levels were equivalent in DT and control recipients (27.0 ± 1.3 vs. 29.6 ± 1.1 mM, ns) (Fig. 2A).
We transplanted 400 BALB/c donor islets under the kidney capsule of recipient mice. To assess the effect of moDCs on early graft function, we compared BG readings 1 day posttransplant between DT and control recipients. In contrast to their pretransplant BG readings, DT recipients had significantly lower BG readings compared to control recipients on day 1 posttransplant (11.0 ± 1.9 vs. 19.1 ± 1.4 mM, p = 0.004) (Fig. 3A). Furthermore, DT recipients had a significantly greater chance of achieving normoglycemia than control recipients (70% vs. 0%, p = 0.002, chi-square test).
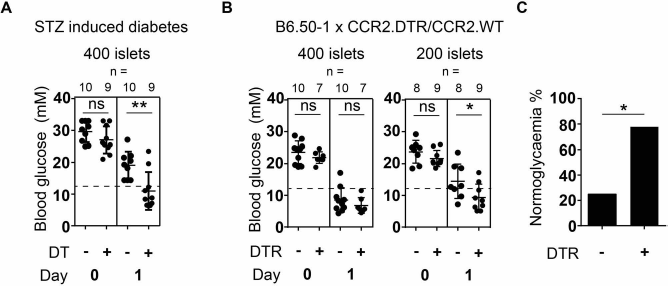
Peritransplant depletion of moDCs in recipients of allogeneic islet transplants results in improved early graft function and a superior probability of achieving normoglycemia. (A) Blood glucose levels in CCR2.DTR recipients of 400 allogeneic BALB/c islets pretransplant and at 1 day posttransplant with or without administration of DT at 3 days and 1 day pretransplant. Recipients were treated with STZ 250 mg/kg 4 days prior to transplantation. (B) Blood glucose levels in B6.50-1 × CCR2.DTR (moDC depleted) or B6.50-1 × CCR2.WT (control) recipients of 200 or 400 allogeneic BALB/c islets pretransplant and at 1 day posttransplant, with the administration of DT at 3 days and 1 day pretransplant. (C) Incidence of achieving normoglycemia (as defined by BG <12 mM) in B6.50-1 × CCR2.DTR or B6.50-1 × CCR2.WT recipients of 200 allogeneic BALB/c islets at 1 day posttransplant as shown in (B). ∗p < 0.05, ∗∗p < 0.01, two-tailed Student's t-test (B), two-tailed chi-square test (C). Error bars indicate SD. Each data point represents an individual recipient. Horizontal dashed line indicates blood glucose of 12 mmol/L. DT, diphtheria toxin; CCR2.DTR, transgenic for the DT receptor under the CCR2 promoter; WT, wild-type; moDCs, monocyte-derived dendritic cells; B6.50-1, a C57BL/6 strain whose transgenic overexpression of a membrane protein in islet β-cells results in spontaneous nonimmune diabetes.
Depletion of moDCs Permits the Use of Lower Islet Masses for Successful Allogeneic Islet Transplantation
Because of continuing issues with lack of donor organs, establishing strategies to reduce the islet mass required to achieve normoglycemia may allow more patients to benefit from islet transplantation. Furthermore, any strategy that improves the chances of achieving normoglycemia could also increase the likelihood of successful long-term outcomes. To determine whether depletion of moDCs could allow for this, we next utilized the B6.50-1 mouse model of diabetes.
B6.50-1 mice bred on the C57BL/6 genetic background spontaneously develop a stable mild to moderate form of nonimmune diabetes due to overexpression of an MHC heavy chain transgene in their islet β-cells, leading to disruption of their secretory pathway 15 . We have previously established that these mice typically require 400 donor islets to reliably achieve normoglycemia post-islet transplant15,17. To avail depletion of moDCs in this model of diabetes, we crossed the B6.50-1 and CCR2.DTR lines and selected double transgenic F1 progeny to act as transplant recipients. These mice exhibit both the B6.50-1 diabetic phenotype as well as the ability for DT-induced conditional depletion of moDCs. Littermates that were B6.50-1 transgenic but DTR WT (i.e., without DTR) were used as control recipients. As shown in Figure 2, DT administration of CCR2.DTR mice resulted in significant depletion of moDCs but had no effect in WT controls.
We first sought to confirm that these B6.50-1 × CCR2. DTR heterozygous recipients could achieve normoglycemia with transplantation of 400 allogeneic islets, as is the case with the parent B6.50-1 line. We administered DT (20 ng/g IP on days −3 and −1) to both groups resulting in depletion of moDCs in DTR recipients. Pretransplant BG levels were similar in both groups (23.6 ± 1.2 vs. 21.9 ± 0.7 mM, ns) (Fig. 3B). We transplanted 400 donor BALB/c islets under the kidney capsule of recipients and assessed graft function by BG measurement 1 day posttransplant. Transplantation of 400 islets successfully achieved normoglycemia in 80% of control recipients. There was no additional benefit in the DTR group in either graft function (6.8 ± 0.9 vs. 8.5 ± 1.2 mM, ns) or incidence of normoglycemia (100% vs. 80%, ns, chi-square test) (Fig. 3B).
We then used a suboptimal donor dose, namely, 200 islets rather than 400. Pretransplant BG levels were again similar between DTR and control recipients (21.7 ± 0.8 vs. 23.8 ± 1.2 mM, ns) (Fig. 3B). With these marginal mass islet transplants, recipients with DT-induced moDC depletion had significantly lower posttransplant BG readings than control recipients (9.3 ± 1.4 vs. 14.5 ± 1.9 mM, p = 0.045) (Fig. 3B). Furthermore, DT-induced moDC depletion resulted in a significantly higher likelihood of achieving normoglycemia than seen in control recipients (78% vs. 25%, p = 0.03, chi-square test) (Fig. 3C).
Depletion of moDCs Results in Improved Graft Survival During the Acute Allograft Rejection Period
Acute islet allograft rejection (T-cell mediated) occurs usually within 2 weeks posttransplant. Since moDC depletion improved early graft function, we next sought to determine whether it could also lead to improvements in graft survival over the acute rejection period. We transplanted 400 donor BALB/c islets into diabetic B6.50-1 × CCR2.DTR or B6.50-1 × CCR2.WT recipients. Both groups received nine injections of DT from day −4 to 15. Successful transplantation was defined by posttransplant BG levels of <2 mM, with graft failure defined as either failure to achieve primary normoglycemia or by two consecutive BG readings >18 mM after an initially successful transplant. The DTR group had significantly longer graft survival compared to the control group (21 days vs. 11 days, p = 0.005) (Fig. 4). Notwithstanding, after cessation of DT administration, reconstitution of moDCs occurred rapidly (not shown) and coincided with the occurrence of graft failure in DTR recipients.

Peritransplant depletion of moDCs in recipients of allogeneic islet transplants results in improved graft survival. Plot of graft survival in B6.50-1 XCCR2.DTR (moDC depleted, n = 4) or B6.50-1 × CCR2.WT (control, n = 6) recipients of 400 allogeneic BALB/c islets, with administration of DT from 3 days pretransplant until 15 days posttransplant. ∗∗p < 0.01, log-rank (Mantel-Cox) test. DT, diphtheria toxin; CCR2.DTR, transgenic for the DT receptor under the CCR2 promoter; WT, wild-type; moDCs, monocyte-derived dendritic cells.
Discussion
Despite much promising work performed by various groups over many years, islet transplantation as a widespread routine therapeutic option for patients afflicted by type 1 diabetes remains a distant dream. The success of the steroid-free Edmonton protocol was a major advance toward this goal 18 ; however, islet transplantation is still beset by major hurdles. Chief among these is the lack of donor organs, which when combined with early islet damage, poor engraftment, suboptimal graft function, and therefore the usual need for more than one transplant, constitutes a major impediment to establishing successful large-scale transplant programs3,4.
Understanding the factors in early posttransplant graft dysfunction should avail the identification of strategies to mitigate the problem and improve outcomes. Various cellular and humoral factors have been implicated in early posttransplant graft dysfunction including IBMIR 3 , macrophages 9 , neutrophils 10 , natural killer (NK)T cells 10 , IFN-γ 8 , IL-1β7,8, and iNOS 7 . In this study, we have identified moDCs as another potential participant in the complex inflammatory posttransplant setting, resulting in adverse effects on graft function and survival.
moDCs are inflammatory dendritic cells that are rare at the steady state and only typically become abundant during infection and inflammation 19 . They have been identified in various inflammatory settings including infection19,20 and autoimmunity 13 . Until recently, experimental studies on moDCs during transplantation have been scant. In one recent study, moDCs rapidly became prominent following experimental heart and kidney allotransplantation 11 . Similarly, we have also observed rapid accumulation of splenic moDCs (within 1 day) following alloantigen exposure in naive mice 12 . In a previous study, also using the CCR2.DTR mouse model, we found that moDCs were capable of educing T-cell differentiation 14 . Some of the factors associated with these moDC functions included iNOS and IFN-γ, both of which may be toxic to pancreatic islets8,21. In this current study, we have found that moDCs also impair early graft function post-allogeneic islet transplantation and that this is associated with the need for transplantation of higher islet masses to achieve normoglycemia. We have also found them to contribute to graft failure during acute rejection, even when a sufficient islet mass is transplanted to allow for initial achievement of satisfactory graft function.
It should be noted that B cells, conventional dendritic cells (cDCs), and neutrophils have no detectable CCR2 13 . Moreover, while a subset of activated T cells express modest levels of CCR2, they are nevertheless not affected by DT injection in CCR2.DTR mice 13 . While the CCR2. DTR model allows for depletion of CCR2+ monocytes, we believe that the bulk of the DT effect is due to depletion of moDCs rather than monocytes. CCR2+ monocytes (the immediate precursors of moDCs) are recruited from the blood to sites of inflammation where they differentiate into moDCs. We have found that, under inflammatory conditions, CCR2+ monocytes readily differentiate into MHCII+CD11c+ moDCs and that at most sites of inflammation and T-cell priming the majority of CCR2+ cells ultimately become moDCs rather than monocytes 13 . Furthermore, up to 30% of monocytes are CCR2− and therefore not subject to DT-mediated deletion (Fig. 2B and C), whereas virtually all moDCs are CCR2+ and so >90% are deleted by DT administration 13 . Interestingly, unlike in CD11c.DTR mice 22 , which may express DTR ectopically in other vital sites (e.g., gut) 23 resulting in the need for irradiation chimeras, we did not observe any toxicity in CCR2.DTR transgenic mice under our regimen.
It has been mooted that peritransplant inflammation could release “danger” signals, which might favor moDC recruitment24,25. Indeed, CCL2 (ligand for CCR2) is expressed in isolated islets26,27 and at the graft site within the day following transplantation26,28. The improved graft function in moDC-depleted recipients as early as the first day posttransplant is strongly suggestive that moDCs can participate in innate immune responses against islet allografts. The initiation of adaptive immune responses, involving T- and B-cell recruitment and activation, would presumably require longer periods of time to become evident. We did find moDCs to also contribute to graft failure at around 11 days posttransplant, compared with moDC-depleted recipients that survived until 21 days posttransplant. The relative contributions of early islet losses versus induction of donor-specific adaptive immune rejection occurring at later time points are currently unclear, but both factors are likely to be involved. Our findings suggest that impaired moDC responses may in part account for the prolongation of islet allograft survival previously observed in recipients genetically deficient in CCR228,29 or CCL2 26 .
Overall, our results indicate that therapeutic strategies aimed at depleting moDCs or impairing their function may allow for successful outcomes and potentially permit transplantation with lower islet masses, potentially assisting in overcoming some of the current problems associated with islet transplantation. Nevertheless, a number of caveats remain regarding these findings. Most importantly, the clinical applicability of the treatments used to deplete moDCs in the experiments described in this study is somewhat uncertain. The use of genetic ablation approaches, equivalent to the CCR2.DTR mouse model, in human transplant recipients is obviously impractical. It would therefore be of significant interest to determine whether other depletion strategies, such as an anti-CCR2-depleting antibody, might also be efficacious in these settings30,31. Alternatively, treatments to impair moDC function (as opposed to induce moDC depletion) could also be studied in the future. A number of CCR2 receptor antagonists have been previously developed and tested in different settings, suggesting that moDC impairment may be a potentially viable strategy32,33. In conclusion, we contend that our findings suggest that further investigation in this area is warranted in order to understand the mechanisms of moDC function, particularly in the peritransplant setting, and to develop potent clinical strategies to target them.
Footnotes
Acknowledgments
The authors thank Merle Dayton, Catherine Yates, and Lauren Wilkins for animal care and technical assistance. This work was supported by the Rebecca L. Cooper Foundation, National Health and Medical Research Council of Australia (NHMRC) grants (1037321, 1043414, 1080321, and 1105209), NHMRC Independent Research Institutes Infrastructure Support Scheme grant (361646), and Victorian State Government Operational Infrastructure Support grant. K.V.C. has received a Kidney Health Australia Biomedical Scholarship and an NHMRC Postgraduate Scholarship. We acknowledge the Wurundjeri people of the Kulin nation as the traditional owners and custodians of the land on which most of the work was performed. The authors declare no conflicts of interest.