
Abstract
It is possible to generate induced pluripotent stem (iPS) cells from mouse and human somatic cells by ectopic expression of defined sets of transcription factors. However, the recommendation that somatic cells should be utilized at early passages for induced reprogramming limits their therapeutic application. Here we report successful reprogramming of human fibroblasts after more than 20 passages in vitro, to a pluripotent state with four transcription factors: Oct4, Sox2, Klf4, and c-Myc. The late passage-derived human iPS cells resemble human embryonic stem cells in morphology, cell surface antigens, pluripotent gene expression profiles, and epigenetic states. Moreover, these iPS cells differentiate into cell types representative of the three germ layers in teratomas in vivo, and directed neuronal differentiation in vitro.
Introduction
The generation of human embryonic stem (hES) cells with the ability to differentiate into all cell types of the body opened up an exciting opportunity for cell-based transplantation therapy for regenerative diseases (5,21). However, an ideal requirement for stem cell-based therapy is to use stem cells derived from patients to circumvent immune rejection of the cells following transplantation. Transfer of a somatic cell nucleus to an enucleated oocyte [somatic cell nuclear transfer (SCNT)] and activation and development of the resulting SCNT embryo to the blastocyst stage embryo allowed derivation of donor-specific embryonic stem cells in rodents and primates, but as yet not in humans.
Recently it has been shown that forced expression of four transcription factors, namely Oct3/4, Sox2, Klf4, and c-Myc (OSKM), in human fibroblasts can reprogram the cells to a pluripotent state. These induced pluripotent stem (iPS) cells exhibit the morphology and cell cycle properties of hES cells, the expression of hES cell marker genes, and formation of teratomas in immunodeficient mice (7,8,17,29,39,44,48). Oct4 and Sox2 have been shown to play a key role in maintaining pluripotency (6,28). c-Myc and Klf4 are two tumor-related factors. c-Myc has been shown to enhance proliferation and transformation (2). It also may induce global histone acetylation (12), thus allowing Oct4 and Sox2 to bind to their specific target loci. Klf4 has been shown to repress p53 expression (43), and p53 protein is related with Nanog suppression during ES cell differentiation (26).
Other combinations of transcription factors, such as Nanog and Lin28 combined with Oct3/4 and Sox2, are also capable of inducing reprogramming of somatic cells (55). In addition to the fibroblasts used in the above mentioned studies, other cell types, including keratinocytes (1,31), liver cells (3,45), lymphocytes (14), neural progenitors/stem cells (11,23), pancreatic β-cells (45), and stomach cells (3), have been reprogrammed by exogenous expression of the four transcription factors. iPS cells have been generated from patients of genetically inherited diseases as an alternative to therapeutic cloning or SCNT for the generation of patient-specific pluripotent stem cells (8,10, 30,37,41).
The source of donor cells is one of the concerns for direct pluripotent reprogramming. Recently we have identified cell types in the mouse that reprogram very efficiently with a reprogramming rate of 1.14% (49). However, typically the generation of iPS cells is still inefficient, ranging between 0.01% and 0.2% when using directed retroviral infection of adult cells with vectors expressing OSKM. Therefore, it is necessary to begin infection with a large amount of cells. Practically, it would be difficult to harvest enough cells from a normal clinical biopsy (~6 mm) in less than three passages, which is the recommended passage for reliable reprogramming (32,38). In this study, we examined whether late passage human fibroblasts could be reprogrammed to pluripotency by the four factors: OSKM. We examined whether late passage-derived human iPS cells resembled human ES cells in morphology, cell surface antigens, pluripotent gene expression profiles, and epigenetic states. In addition, we investigated the ability of these iPS cells to differentiate randomly in vivo into cells representative of the three germ layers in teratomas, and by directed neuronal and neural crest differentiation in vitro.
Materials and Methods
Animals
All mice were housed at the Monash Medical Centre Animal Facilities, Clayton, Australia. SCID mice were housed in specific pathogen-free (SPF) conditions. All animal experimentation was conducted with protocols and ethics approved by the Monash Medical Centre Animal Welfare Committee.
Cell Culture
Human fibroblast Detroit 551 (D551) cells were obtained from ATCC (www.atcc.com). Retroviral packaging Platinum-A (Plat-A) cells, a transfected human embryonic kidney (HEK)-293T cell line, were obtained from Cell Biolabs, Inc. (www.cellbiolabs.com). Mouse embryonic fibroblasts (MEFs), D551 cells, and Plat-A cells were cultured in Dulbecco's modified eagle medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen). Human embryonic stem (hES) cell line, MEL-1, was obtained from Australia Stem Cell Centre (ASCC) and cultured in DMEM supplemented with 20% FBS (Hyclone), 1 mM L-Gluta-max, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol, 1% ITS, and 10 ng/ml bFGF (all from Invitrogen). Human iPS cells were cultured in the medium described above for MEL-1 cells with changing culture medium daily. Human iPS cell lines, ES4CL1-4 and MR90, were kindly provided by Prof. J. Thomson (Genome Center of Wisconsin, Madison, WI) and used in the study.
For passaging of the cells, either mechanical dissociation or enzymatic digestion was used. Mechanical dissociation was performed to dissect iPS cell colonies into smaller cell clumps using a 1-ml insulin syringe with a 29-gauge needle, and then transfer them to a fresh MEF layer. Enzymatic dissociation was performed by incubating the iPS cell culture with 4 mg/ml dispase (Invitrogen) at 37°C for 10 min. The detached colonies were pipetted up and down to dissociate them into small pieces, and then centrifuged at 300 × g for 5 min. The pellet was resuspended and cultured on a fresh MEF layer in the hES cell medium containing 10 mM Y-27632 (Calbiochem), a ROCK inhibitor (52), for 24 h, before returning to standard hES cell medium.
Feeder Preparation
MEFs were isolated from day 13.5 postcoitum fetuses of CD1 mice. They were treated with 10 μg/ml mitomycin C (Sigma) for 3 h to arrest mitosis, then washed in PBS, and replated at a density of 4 × 104 cells/cm2 on gelatin (Sigma)-coated tissue culture dishes.
Retroviral Production and iPS Cell Generation
Moloney-based retroviral vectors (pMXs) containing the human cDNAs of OCT4, SOX2, KLF4 and c-MYC were obtained from Addgene (www.addgene.org). Nine microgram of each plasmid was transfected into viral packaging Plat-A cells using Fugene 6 (Roche). Virus-containing supernatants were collected 48 and 72 h post-transfection and filtered through a 0.45-μm pore size filter and supplemented with 4 μg/ml of polybrene (Sigma). D551 cells at passage 23 were plated 24 h prior to infection at a density of 1 × 104 cells/cm2. Retroviral supernatants of four transcription factors were mixed in equal quantities and added to the target cells at 24 and 48 h. The culture medium for the infected cells was changed to hES cell medium at day 4 postinfection. The cells were maintained in the culture with medium refreshment every day up to about 4 weeks or until the cells reached over confluence, in which case the cells were replated on a MEF feeder layer. To establish iPS cell lines, iPS cell colonies were picked-up based on hES cell-like colony morphology at about 4 weeks postinfection. The picked colonies were expanded on fresh MEF feeder layer in hES cell medium. To estimate transduction efficiency of D551 cells, pMXs-GFP retroviral vectors were also transfected to Plat-A cells using the same method as described above. pMXs retroviruses containing the GFP cDNA were added to D551 cells. The number of cells expressing GFP was evaluated by flow cytometry 48 h after infection.
FACS Analysis
Cells were dissociated with 0.25% trypsin-EDTA (Invitrogen) for 5 min. Resuspended cells were filtered through a 40-μm cell strainer (BD Falcon) and analyzed using a BD FACSCanto flow cytometer (BD).
Karyotype Analysis
Karyotype analysis was performed in the Cytogenetics, Monash Medical Centre. Briefly, the cells were grown to subconfluency at which point colcemid (Invitrogen) was added to a final concentration of 0.1 mg/ml for 30–60 min. Cells were then trypsinized, pelleted by centrifugation at 1,200 rpm for 5 min, resuspended in 2 ml of hypotonic solution (0.56% KCl + 0.5% Na citrate), and incubated for 30 min in a 37°C water bath. After pelleting of cells again by centrifugation, the supernatant was removed and ice-cold fresh fixative comprised of one part acetic acid to three parts methanol was added drop-wise to a final volume of 10 ml. Cells were fixed for 10 min at −20°C, pelleted by centrifugation, supernatant was removed, and 2 ml fresh fixative added. Metaphases from cells prepared in this manner were assessed by G-banding. At least 20 metaphase spreads were evaluated.
RT-PCR and PCR
Total RNA was isolated using RNeasy kit (Qiagen) and cleaned using DNA-free kit (Ambion) followed by cDNA synthesis using Superscript III Reverse Transcriptase and Oligo (dT) primers (Invitrogen). Genomic DNA was isolated using DNeasy Blood & Tissue kit (Qiagen). PCR was performed with Taq DNA polymerase (Invitrogen). Primer sequences are supplied in Table 1.
Primers for RT-PCR and PCR Reactions

Alkaline Phosphatase and Immunofluorescence Staining
Cells were fixed in 4% paraformaldehyde (Sigma) in PBS for 20 min at room temperature followed by three washes with PBS. Alkaline phosphatase (AP) staining was performed using the Alkaline Phosphatase Detection Kit (Millipore), according to the manufacturer's instructions. For immunofluorescence, cells were blocked with 10% goat serum (Sigma) in PBS/0.1% Triton X and incubated with primary antibodies overnight at 4°C. Secondary antibodies were incubated for 1 h at room temperature. The primary antibodies included pluripotent stem cell makers: Oct4 (Santa Cruz Biotechnology), SSEA3, SSEA4, Tra-1-60, Tra-1-81 (all from Millipore), and neural differentiation makers: β-tubulin (Millipore), Pax-6 (Developmental Studies Hybridoma Bank), p75 (Sigma), and Peripherin (Sigma). Appropriate Alexa Fluor® 594 or 488 conjugated secondary antibodies (Invitrogen) were used to visualize immunoreaction. Nuclei were counterstained with 1 μg/ml of Hoechst-33342 (Sigma-Aldrich) in some slides. The negative control was performed following the whole procedure described above except replacing the primary antibodies with blocking solution.
Bisulfite Genomic Sequencing
Genomic DNA (1 μg) from human iPS cells, MEL-1 cells, and D551 cells were processed for bisulfite modification using CpGemone DNA modification kit (Millipore). Two conserved CpG-enriched regions in the Oct4 promoter and one conserved CpG-enriched region in the Nanog promoter were selected to be amplified by PCR (13). The PCR products were subcloned into pGEMT-easy vector (Promega). Ten clones of each sample were verified by sequencing with Sp6 universal primer and the methylation status was determined using BiQ Analyzer software (4). Primers used for PCR amplification were provided in Table 1.
Teratoma Formation
Human iPS cells grown on MEF feeder layers were dissociated by dispase treatment, centrifuged, and injected into hind leg muscle of diabetes severe-combined immunodeficient (NOD-SCID) mice. Teratoma tissues were collected 6–8 weeks postinjection, and processed for paraffin embedding and hemaoxylin and eosin (H&E) staining following standard procedures.
Neurosphere Formation and Neuronal Induction
The human iPS cell lines, AUS-1, ES4CL1-4, and MR90 (50,55) were treated with 500 ng/ml of Noggin (R&D) for 14 days to induce their differentiation toward neuroectoderm, as described elsewhere (9,40). Noggin-treated cells were mechanically cut into small pieces, and each piece was then further subcultured in 96-well plates in neural basal media (NBM) supplemented with basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF) (20 ng/ml each, R&D) to induce neurosphere formation. We plated 24 pieces in 24 wells in 96-well plates for each cell line in one experiment. We performed at least two replicates. Medium was changed every second day. The frequency of neurosphere formation was measured after 7 days of culture as determined by spherical cluster of cells showing presence of rosette-like structures. After 14 days of culture in suspension, the iPSC-derived neurospheres were plated onto laminin-coated dishes in NBM for 5 days to induce their differentiation towards neurons (42). Differentiation towards neural crest-like cells was performed using previously published protocols (16). Briefly, iPS-derived neurospheres were plated onto a feeder layer of MEFs in NBM supplemented with bFGF and EGF (20ng/ml each). After 24 h, cultures were treated with the small molecule Rho/ROCK inhibitor Y27632 at a final concentration of 25 μM. After 7 days, cultures were fixed with 4% paraformaldehyde and processed immunostaining.
Results
In this study, we investigated generation of human iPS cells from a human fibroblast line, Detroit 551 (D551), at passage 23. D551 fibroblasts have a finite life span of about 25 serial passages from the tissue of origin. We used Plat-A cell line as a potent retrovirus packaging cell line. We transfected Plat-A cells with retroviral vector pMXs containing open reading frame of human transcription factors Oct3/4, Sox2, Klf4, and c-Myc. We collected and introduced the retroviruses containing the four factors into the D551 cells. We infected pMX-GFP-expressing retroviruses into the D551 cells to monitor the viral infection efficiency (Fig. 1A). The single-factor infection efficiency was about 24.5%, as determined by GFP expression in the cells by FACS analysis. The cells were refreshed with hES medium 4 days after infection. Nine days following infection, the fibroblasts displayed primary morphological changes with cell clumping. By 19 days following infection, two types of cell colonies, which resembled hES cell colony morphologies with a high nucleus/cytoplasm ratio (Fig. 1B), could be found in the cultured cells. Type 1 colonies were comprised of multilayers of compacted cells and type 2 colonies displayed a single layer of cells. Both types of colonies could grow to large colonies and be picked up mechanically and expand on fresh MEF feeder layers at around 4 weeks after infection. The expanded ES-like iPS colonies exhibited a flat and tightly compacted morphology with distinct edges (Fig. 1B), and the cells exhibited a large nucleus with prominent nucleoli (Fig. 1B), similar to MEL-1 cells, a human ES cell line, cultured in our laboratory (27). The cells were passaged every 4–6 days, which is similar in growth rate to the MEL-1 cells. Three hES cell-like iPS cell lines were expanded and named AUS-1, -2, and -3, respectively.

Generation of iPS cells from passage 23 D551 cells. (A) D551 cells at passage 23 were introduced with pMXs retroviruses containing GFP cDNAs. (A-a) Phase contrast image of D551 cells; (A-b) shows the images of GFP fluorescence of the same cells, and (A-c) is the merged image of A-a and A-b. (B) Progression of cell morphology changes from fibroblasts to iPS cells. (a) D551 cells at day 1 when receiving retroviruses containing 4 reprogramming factors. (b, c) 19 days after infection, type 1 and type 2 hES cell-like colonies (dotted areas), respectively. (d) 26 days after infection, typical image of hES cell-like colony. (e) AUS cells at passage 1 on the MEF feeder cells by mechanical dissociation. The image was taken at day 2 after passaging. (f) AUS cells at passage 2,;the image was taken at day 4 after passaging. (g) AUS cells at passage 4 by Dispase dissociation. The image was taken at day 2 after passaging. (h) AUS cells at passage 4 in higher magnification (40×). Scale bars: 200 μm.
We first characterized the AUS cells to test the expression of pluripotent stem cell markers. All three expanded AUS cell lines were positive for the expression of AP, Oct3/4, SSEA3, SSEA4, TRA-1-60, and TRA-1-81 (Fig. 2A).

Characterization of AUS cells. (A) AUS cells were positive for AP, Oct4, SSEA3, SSEA4, TRA-1-60, and TRA-1-81. Scale bars: 200 μm. (B) RT-PCR showed that induced human iPS cell lines (AUS-1, AUS-2, and AUS-3) expressed endogenous human ES cell marker genes, such as Oct4, Sox2, Nanog, and Rex1. Exogenous Oct4, Sox2, and c-Myc genes were effectively silenced, while the exogenous Klf4 transgene was still expressed. Endo, endogenous; Exo, exogenous. (C) Integration of Oct4, Sox2, Klf4, and c-Myc transgenes in the genome of AUS cell lines was confirmed by genomic DNA PCR using transgene-specific primers, with D551 and MEL-1 cells as negative controls, and the respective vector as a positive control.
RT-PCR analysis showed that the AUS lines expressed endogenous Oct4, Sox2, and Nanog pluripotent marker genes, which were not detectable in the parental D551 cells (Fig. 2B). Another pluripotent marker, Rex1, was detectable in AUS-1 line, and weakly detected in AUS-2 and AUS-3. The MEL-1 cells, the AUS cell lines, and their parental D551 cells all expressed endogenous c-Myc and Klf4. We examined the persistent expression of viral transgenes by RT-PCR and showed that viral Oct4, Sox2, and c-Myc transgenes were efficiently silenced; however, viral Klf4 transgene was still expressed in the AUS cells. Genomic DNA PCR showed that all AUS lines harbored integrated transgenes of four transcription factors (Fig. 2C).
We investigated the DNA methylation status of CpG dinucleotides in the Oct4 and Nanog promoter regions in the AUS lines. Two CpG-enriched regions in Oct4 promoter and one CpG-enriched region in Nanog promoter were selected for analysis. Bisulphate genomic sequencing analysis showed that both the Oct4 and the Nanog promoter regions were demethylated in the AUS cells, similar to human ES cells (MEL-1), whereas the same regions were highly methylated in parental D551 cells (Fig. 3).

Bisulfite sequencing analysis of Oct4 and Nanog methylation in AUS-1, MEL-1, and D551 cells. Top numbers indicate the CpG position relative to the transcription start site. Global percentages of methylated cytosines (% Me) are shown. Each row of circles for a given amplicon represents the methylation status of each CpG in one bacterial clone for the region. Ten clones are shown. Open and filled circles indicate unmethylated and methylated CpG dinucleotides, respectively.
We performed karyotype analysis on two AUS cell lines and the result showed that both lines maintained a normal karyotype (46, XX) (Fig. 4A, B).
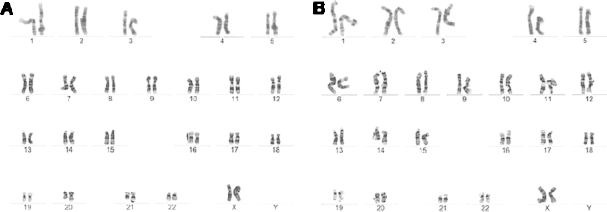
AUS-1 (A) and AUS-2 (B) cells have a normal karyotype (46, XX) at passage of 10 by chromosomal G-banding analysis.
We evaluated the in vivo differentiation capacity of AUS cells by teratoma formation. AUS cells formed teratomas after intramuscular injection into hind legs of NOD-SCID mice. H&E staining of the teratomas revealed that teratomas contained three germ layer tissues, including secretary gland (endoderm), epidermal tissue (ectoderm), and cartilage (mesoderm) (Fig. 5a–c).

Hematoxylin and eosin staining of teratoma from AUS cells showed (a) keratinized epithelium (arrows), (b) cartilage, and (c) secretary epithelium. In in vitro differentiation system, AUS cells formed embryoid bodies after 7 days in suspension culture (d), further differentiated to a cyst-like structure (arrow) after 5 days of plating EBs on petri dishes (e). Scale bars: 200 μm.
We then evaluated the in vitro differentiation capacity of all three AUS cell lines by formation of embryoid bodies (EBs) in suspension culture. All the AUS cells formed EBs after 7 days of culture (Fig. 5d) and differentiated into different cell types and structures (Fig. 5e) after attachment for 5 days. To evaluate the in vitro-directed differentiation capacity of AUS cells, we applied a directed neuronal differentiation protocol to induce the AUS-1 cells to neuronal lineage. Similar to hES cells (data not shown), AUS cells were responsive to Noggin treatment and able to differentiate into neuroectoderm, as shown by expression of Pax6 (Fig. 6a). The Noggin-treated cells could form neurospheres in the presence of bFGF and EGF in suspension culture (Fig. 6b). The average neurosphere formation rates (mean ± SEM%) were: AUS-1 cells: 56.1 ± 12.6 (n = 3), ES4CL1: 80.5 ± 13.8 (n = 2), ES4CL2: 41 ± 1.9 (n = 2), ES4CL3: 62.5 ± 4.2 (n = 2), ES4CL4: 23.3 ± 6.7 (n = 2), MR90C2: 87.5 ± 8.3 (n = 2), and MR90C4: 87.5 ± 8.3 (n = 2). Efficiency of neurosphere formation from AUS-1 cells was similar to that from other human iPS cell lines (Fig. 6d). Further, differentiation toward neurons was achieved by culturing neurospheres onto laminin-coated plates. βIII-Tubulin expression was detected in the differentiated cells (Fig. 6c). Neurospheres derived from AUS-1 cells were induced toward neural crest-like cells by coculturing them on MEF feeder layers with the addition of Y27632. Similar to what has been published for hES cells, migratory progenitors are observed in the neural crest induction cultures that express p75 (Fig. 6e) and can differentiate to neurons expressing the peripheral marker, Peripherin (Fig. 6f).
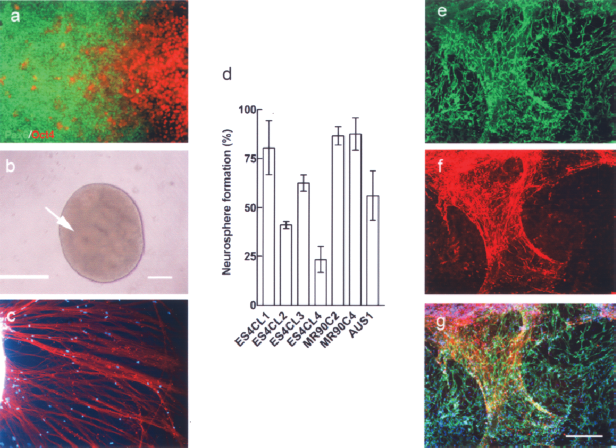
In vitro-directed differentiation of AUS-1 cells to neuronal cells and neural crest. (a) AUS-1 cell colonies were treated with Noggin for 14 days to induce differentiation toward neuroectoderm, as shown by expression of Pax6+ (green); undifferentiated Oct4+ cells (red) are also observed. (b) Neurospheres formed in suspension culture for 5 days. Neruospheres are morphologically characterized by containing rosette-like structures formed by clustering of neural stem cells (arrow). (c) Neurospheres plated on laminin substrate differentiated to neurons in 7 days, as shown expressing of βIII-tubulin. (d) Graph showing efficiency of neurosphere formation for Noggin-treated AUS-1 iPS cells compared with six other Noggin-treated iPS cell lines. (e) AUS-1-derived neurospheres cocultured on a MEF feeder layer, following Y27632 treatment, results in their differentiation to migratory p75+ progenitors, indicative of neural crest-like cells. (f) A subset of p75+ cells further differentiated to neurons expressing the peripheral neuronal marker, Peripherin. (g) Merged image of (e) and (f) with DAPI counterstaining. Scale bars: 200 μm.
Discussion
In summary, the late passage human fibroblasts could be reprogrammed to a pluripotent state by retroviral transduction of Yamanaka four transcription factors, albeit with a low infection efficiency. The iPS cells, named AUS cells, were characterized and shown to share many similar properties to hES cells, including morphology, cell surface markers, gene expression profiles, promoter methylation status, EB formation, teratoma formation, and directed neuronal differentiation capacity. The endogenous self renewal genes (Oct4 and Nanog) were reactivated and three of the transgenes, which included c-Myc, were silenced.
It has been highly recommended that target cells for iPS cell derivation be used within three passages in order to avoid replicate senescence (38,47). The reported efficiency of iPS cell derivation is extremely low, ranging between 0.01% and 0.2% when using direct viral infection of adult cells with vectors expressing the Yamanaka four reprogramming factors (33,48,54). Practically, it is difficult to harvest enough cells for iPS cells derivation from a 6-mm human skin biopsy in less than three passages. In this study, we attempted to generate human iPS cells from late passage human fibroblasts. Detroit 551 (D551) cells are human fetal skin fibroblasts, which have a finite life span of about 25 serial passages from the tissue of origin as stated in the ATCC catalogue. We used cells at passage 23 as target cells for infection by retroviral delivery of the four reprogramming factors. The infection efficiency was monitored by retroviral infection of the cells with pMX-GFP. The low infection rate (24.5%) might reflect that the D551 cells were not actively proliferating at passage 23, because retroviral infectivity is limited to dividing cells (35). Human ES-cell like cells, appearing as colonies, were recognizable from day 19 after two rounds of viral infection in 48 h. The colonies could be picked up manually at around day 26 and propagated on MEF feeder cells. The cells were demonstrated as iPS cells with a normal karyotype by a series of characterization assays.
Unlike the 293T cells used in most other studies to produce human iPS cells, Plat-A cells were used as retrovirus packaging cells in the present study. Plat-A cells were generated by stably transfecting two unique packaging constructs into 293T cells (24,34). The two packaging constructs use the EF1α promoter to express gagpol, or amphotropic env genes. Plat-A cells produce high-titer retroviruses (1 × 106 IU/ml) by transient transfection of pMXs vectors (24). Murine Molony Leukemia Virus (MMLV)-based retroviral vectors are known to undergo transcriptional silencing in pluripotent cells. RT-PCR-specific for retroviral trangene Oct4, Sox2, and c-Myc transcripts confirmed the effective silencing in AUS cells. However, silencing was not observed for the KLF4 transgene. It has been reported that iPS cells made by retroviral infection often maintain viral gene expression (8,39), which is one of the drawbacks in the use of retroviruses. Therefore, new approaches to deliver reprogramming factors to the target cells, especially non-integrated methods, need to be investigated and optimized (18,19,36,46,53,56,57).
AUS cells could form teratomas and differentiate into tissue representatives of the three germ layers when injected into immunodeficient mice. Furthermore, when maintained in suspension culture conditions, AUS cells could form EBs, demonstrating their random differentiation capacity in vitro. Moreover, the AUS cells could be directly differentiated in vitro into neurons and neural crest-like progenitors, confirming that iPS cells are a possible source of personalized pluripotent stem cells for providing therapies in the field of regenerative medicine. We have previously published an efficient system for generation of neural crest-like cells from human embryonic stem cells (16). This protocol involves coculture of neurospheres on a feeder layer of mouse embryonic fibroblasts followed by treatment of the small molecule Rho/ROCK inhibitor, Y27632. The migratory progenitors express neural crest markers, including p75, Sox10, and HNK1. Furthermore, neural crest-like progenitors derived from this system demonstrated differentiation to peripheral neurons in vitro and in vivo. This same protocol was applied to neurospheres derived from AUS1 iPS. Similar to what we have published for hESC, migratory progenitors are observed in the neural crest induction cultures that express p75 and can differentiate to neurons expressing the peripheral maker, Peripherin. Taken together, these properties are characteristic of neural crest, thereby suggesting the generation of neural crest-like progenitors from AUS-1 iPS.
Mouse and human iPS cells have the same capabilities as ES cells isolated from early mammalian embryos—they can self-renew, and are capable to give rise to all tissue types of the body. In contrast to ES cells derived from embryos produced by fertilization, iPS cells can be produced from adult cells from any individual and therefore their availability can circumvent the ethical issues associated with ES cells and circumvent the immune rejection. Somatic cell nuclear transfer is another method to generate patient-matched pluripotent stem cells by introducing a donor nucleus to an enucleated oocyte. The strong advantages of direct reprogramming are the ability to generate patient-specific cell lines with relative ease and without sacrificing human oocytes and embryos. Various attempts have been performed to study the mechanism of induced pluripotency, including the use of small molecule chemicals, such as valproic acid (VPA) (17); the use of different cell types as starting populations (22,23); the use of proteins of four reprogramming factors (57); and suppression of the p53 network to enhance the production of iPS cells (15,20, 25,33,51).
In summary, we demonstrate that human iPS cells can be generated from cells approaching replicative senescence and that these cells closely resemble human ES cells in morphology, growth characteristics, gene expression, and methylation profiles. Also, the AUS cells can be differentiated both in vitro and in vivo, indicating pluripotency. Importantly, when subjected to directed differentiation conditions developed for human ES cells, the human iPS cells can differentiate to cells of the neuronal lineage, including definite neuro-ectoderm, neurospheres that could be differentiated to neurons and neural crest-like progenitors for the first time.
Footnotes
Acknowledgments
The authors are grateful to Prof. J. Thomson for providing the iPS lines MR90 and ES4CL1-4. This work was supported by VIC/NSW government grand and Friedreich Ataxia Research Association (Australasia and USA).