
Abstract
Cellular senescence induces changes in cellular physiology, morphology, proliferative capacity, and gene expression. Stem cell senescence might be one of the major issues of limited efficacy of stem cell transplantation. In this study, we demonstrated that implantation of human umbilical cord mesenchymal stem cells (hUCMSCs) cultured in human umbilical cord serum (hUCS) significantly enhanced neuroplasticity and angiogenesis in stroke and ischemic limb models. Immunophenotypic analysis indicated that hUCMSCs cultured in hUCS had more small and rapidly self-renewing cells than those expanded in FCS. The main cause of greater senescence in FCS-cultured cells was increased generation of reactive oxygen species (ROS). Proteome profiling showed significantly more senescence-associated vimentin in FCS-cultured hUCMSCs than in hUCS-cultured hUCMSCs. In contrast, there was significant upregulation of heat shock protein 27 (Hsp27) in the hUCS-cultured hUCMSCs. By gene targeting, we found that overexpression of Hsp27 may downregulate vimentin expression through inhibition of the nuclear translocation of p65 (NF-κB signaling). Thus, an interaction between Hsp27 and vimentin may modulate the degree of senescence in hUCS- and FCS-cultured hUCMSCs. In summary, hUCMSCs exhibiting senescence are detrimental to cell engraftment and differentiation in animal models via activation of NF-κB pathway. Human stem cells incubated in hUCS might reduce the senescent process through upregulation of Hsp27 to increase implantation efficiency.
Keywords
Introduction
Cellular senescence is the limited ability of primary human cells to divide when cultured in vitro. The molecular mechanism of senescence is not known, although possible causes include oxidative damage, genomic instability, genetic reprogramming, and cell death (19). Senescence-associated alteration of the function and expression of cytoskeletal proteins such as vimentin is still not completely understood. It has, however, been demonstrated that aged cells and cells undergoing replicative senescence have diminished heat shock response and a higher incidence of death when subjected to severe stress (26). Because heat shock protein 27 (Hsp27) is thought to protect cells from oxidative stress, alterations in the production of Hsp27 can further promote aging by increasing the accumulation of damaged proteins.
Mesenchymal stem cells (MSCs) obtained from bone marrow stroma and human umbilical cord matrix are attractive candidates for clinical use in stem cell therapy because they can enhance neurogenesis and angiogenesis in stroke and ischemic limb models (7,21). However, fetal calf serum (FCS) induces anaphylactic reactions (39) and poor cellular engraftment (16). If MSCs are to be used in cell transplantation in humans, it is important to minimize these risks, which result from the use of nonhuman sera in the culture system (4,15,24).
In this study, we applied human umbilical cord serum (hUCS) to human umbilical cord mesenchymal stem cell (hUCMSC) culture to investigate whether hUCS could significantly reduce cell senescence (1), enhance hUCMSCs' proliferative ability and differentiation potential, and improve tissue plasticity in stroke and ischemic limb models compared with hUCMSCs cultured in FCS. In addition, the molecular mechanism of replicative senescence in this cellular system was investigated by studying the target proteins regulated through the NF-κB pathway.
Materials and Methods
Human Umbilical Cord Blood Serum and Umbilical Cord Mesenchymal Stem Cell Preparation
Human umbilical cord blood (hUCB) was harvested in 50-ml Falcon tubes without anticoagulant and processed within 24 h. Protocols for sampling hUCB were approved and provided by StemCyte. Protocols for sampling hUCB were approved by the Institutional Review Board of China Medical University Hospital, Taichung, Taiwan. Written informed consent was obtained from all mothers before labor. Human umbilical cord serum (hUCS) was obtained from hUCB by centrifugation (3,000 rpm, 10 min) (Beckman) as previously described with modification (41). Then, hUCS was boiled at 56°C for 30 min, passed through a filter (0.22 μm, Stericup, Millipore), and diluted with phosphate-buffered saline (PBS) for further application.
Human umbilical cord samples (20 cm length, 20 g weight) were collected and processed for primary culture of human umbilical cord mesenchymal stem cells (hUCMSCs) as previously described (7).
Proliferation Analysis and Immunophenotyping
After hUCMSCs had migrated from the explants, they were passaged into the following media: (i) serum-free DMEM medium (Gibco, BRL), (ii) medium containing 10% FCS, (iii) medium containing only 10% hUCS (23), medium containing 10% hUCS plus 10 ng/ ml epidermal growth factor (EGF) and 10 ng/ml basic fibroblastic growth factor (bFGF) (hUCS+). To test the proliferative potential of hUCMSCs in each cultural protocol, hUCMSCs were plated at 50 and 500 cells/cm2 initially and expanded in the medium to count cell density (yield) for 7 consecutive days. The immunophenotyping procedure was as previously described (7).
In Vitro Differentiation Assay for hUCMSCs
hUCMSCs were passaged and cultured to confluence, and shifted to osteogenic medium, adipogenic medium, and neurogenic medium for differentiation as previously described (7).
Analysis of Intracellular Generation of Reactive Oxygen Species (ROS)
To determine ROS levels, the fluorescence intensity of intracellular staining of 2′,7′-dichlorofluorescein diacetate (DCF-DA) was measured by flow cytometry as previously described (3). In brief, DCF-DA (Sigma-Aldrich) was solubilized in ethanol at a concentration of 10 mM. Cells were inoculated at a density of 2 × 104 cells in 6-cm plastic plates, each of which contained 5 ml of FBS- or hUCS-supplemented MEM, and cultivated overnight. After removal of the medium, the cells in each plate were washed with 2 ml of PBS and incubated at 37°C for 24 h in 5 ml of FBS- or hUCS-supple-mented MEM. After removal of the medium and washing with 2 ml of PBS, they were incubated at 37°C for 30 min in 2 ml of Earl's solution containing 10 mM DCF-DA. After removal of the medium, the cells were washed with 2 ml of PBS. One milliliter of 0.25% trypsin was added, and aspirated out. Cells were then incubated at 37°C for 3 min, suspended in 4 ml of PBS, and transferred into a centrifuge tube. The fluorescence intensity of DCF (the deesterified, oxidized product of DCF-DA) in cells was determined using a flow cytometer.
Animal Brain Ischemia/Reperfusion Model
Adult male Sprague-Dawley rats (weight 250-300 g) were subjected to three-vessel ligation. All surgical procedures were performed using sterile/aseptic techniques in accordance with institutional guidelines (36). The Ethical Committee for animal research at China Medical University Hospital reviewed and approved all animal experiments. The rats were anesthetized with chloral hydrate (0.4 g/kg, IP). Ligation of the right middle cerebral artery (MCA) and bilateral common carotids (CCAs) was performed by methods described previously with modifications. The bilateral CCAs were clamped with nontraumatic arterial clips. Using a surgical microscope, a 2 × 2-mm craniotomy was performed where the zygoma fuses with the squamosal bone. The right MCA was ligated with 10-0 nylon suture. Cortical blood flow was measured continuously with a laser Doppler flowmeter (PF-5010, Periflux system, Perimed AB) in anesthetized animals. A burr hole (1-mm diameter) was made in the right frontoparietal region to allow placement of photodetectors. A probe (0.45 mm in diameter) was stereotaxically placed in the cortex (1.3 mm posterior, 2.8 mm lateral to bregma, and 1.0 mm below the dura). After 90 min of ischemia, the suture on the MCA and the arterial clips on CCAs were removed to allow reperfusion. Core body temperature was monitored with a thermistor probe and maintained at 37°C with a heating pad during anesthesia. After recovery from anesthesia, body temperature was maintained at 37°C with a heat lamp.
Intracerebral Transplantation of hUCMSCs
Cells were cultured in DMEM (Gibco-BRL) with 10% FCS or hUCS at 37°C in a humidified atmosphere of 5% CO2/95% air and antibiotics. Prior to transplantation, cells were incubated with 1 μg/ml bis-benzimide (Hoechst 33342; Sigma), for 5 h at 37°C. One week after brain ischemia, rats were anesthetized with chloral hydrate (0.4 g/kg, IP) and then injected stereotactically with approximately 1 × 106 cells in 3-5 μl PBS as previously described (36). Labeled cells were then collected and washed in PBS three times. Nucleated hUCMSCs were counted using a cytometer to ensure an adequate cell number for transplantation. One week after brain ischemia, rats were anesthetized with chloral hydrate (0.4 g/kg, IP) and then injected stereotactically with approximately 1 × 106 cells in a 3-5 μl PBS suspension through a 26-gauge Hamilton syringe into three cortical areas adjacent to the right MCA, 3.0-5.0 mm below the dura. The approximate coordinates for these sites were l.0-2.0 mm anterior to bregma and 3.5–4.0 mm lateral to the midline, 0.5–l.5 mm posterior to bregma and 4.0–4.5 mm lateral to the midline, and 3.0–4.0 mm posterior to bregma and 4.5–5.0 mm lateral to the midline. The needle was retained in place for 5 min after each injection and a piece of bone wax was applied to the skull defects to prevent leakage of the injected solution. The experimental rats did not receive any immunosuppression.
Neurological Behavioral Measurement
Behavioral assessments were performed 5 days before cerebral ischemia, and 7, 14, and 28 days after cell transplantation. Tests measured body asymmetry, locomotor activity, and grip strength as previously described (36). An elevated body swing test was used to assess body asymmetry after MCA ligation and evaluated quantitatively. Initially, animals were examined for lateral movement when their bodies were suspended by their tails 10 cm above the ground. The frequency of initial head swing contralateral to the ischemic side was counted in 20 continuous tests and was normalized by the baseline score. Rats were subjected to VersaMax Animal activity monitoring (Accuscan Instruments) for about 2 h to record motor activity. This instrument contained 16 horizontal and 8 vertical infrared sensors. The vertical sensors were situated 10 cm from the floor of the chamber. Motor activity was counted as the number of beams broken by the rat's movement in the chamber. Three vertical items were calculated over 2 h: vertical activity, vertical time, and number of vertical movements. Grip strength was analyzed using Grip Strength Meter (TSE-Systems, Germany). The grip strength ratio of each forelimb was measured separately and was calculated as the ratio between the mean strength out of 20 pulls of the side contralateral to the ischemia and the ipsilateral side. In addition, the ratio of grip strength posttreatment (post-Tx) and pretreatment (pre-Tx) was calculated; the changes were presented as a percentage of the pretreatment value.
Immunohistochemical Assessment of Brain Tissue
Animals were anesthetized with chloral hydrate (0.4 g/kg, IP) and their brains fixed by transcardial perfusion with saline, followed by perfusion with and immersion in 4% paraformaldehyde. Immunohistochemical analysis using laser scanning confocal microscopy was performed as previously described (36).
[18F]Fluoro-2-deoxyglucose Positron Emission Tomography (FDG-PET)
To assess the metabolic activity and synaptic density of brain tissue, experimental rats were examined using microPET scanning of [18F]fluoro-2-deoxyglucose (FDG) to measure relative brain metabolic activity as previously described (36).
Mouse Hindlimb Ischemia and Laser Doppler Imaging Analysis
C57BL/6 mice were anesthetized with chloral hydrate (0.4 g/kg, IP). The femoral artery ligation, limb blood flow measurement by laser doppler perfusion imaging (LDPI) (Moore Instruments), and immunohistochemical studies were performed as previously described (22). Under sedation, 12-week-old mice underwent surgical removal of right femoral artery (proximal to the origin) to create unilateral hindlimb ischemia. At 1 day after surgery, mice were performed for cell injection (1 × 106 cells in 300 μl saline) into six separate injections sites of femoral biceps muscle, abductor muscle, semitendinous muscle, semimembranous muscle, sartorius muscle, and adductor muscle.
Proteomic Analysis
Differential protein expression in hUCMSCs plated at 500 cells/cm2 for 7 days through 4 passages in 10% FCS and hUCS was investigated by proteomic analysis. Proteomic assays including 2D gel electrophoresis, silver stain with imaging analysis, in-gel tryptic digestion with peptide extraction, and protein identification by QSTAR XL MS/MS System (Applied Biosystems) were performed according to the manufacturer's recommendations as previously described with modification (40).
Activated Protein Kinase Assay
Western blot analyses of signal transduction protein expression from hUCMSCs were performed after cells were cultured in FCS or hUCS as previously described (7). Briefly, hUCMSCs incubated in FCS or hUCS were lysed in a buffer containing 320 mM sucrose, 5 mM HEPES, 1 μg/ml leupeptin, and 1 μg/ml aprotinin. Lysates were centrifuged at 13,000 x g for 15 min. The resulting pellet was resuspended in sample buffer (62.5 mM Tris-HCl, 10% glycerol, 2% SDS, 0.1% bromophenol blue, and 50 mM DTT) and subjected to SDS-PAGE on 4–12% polyacrylamide gels. The proteins separated in the gel were then transferred to a Hybond-P nylon membrane and incubated with appropriately diluted antibodies to p-Stat3 (1:200; Cell Signaling), Stat3 (1:200; Santa Cruz), p-Akt (1:200; Calbiochem), Akt (1:300; Calbiochem), p-ERK1/2 (1:200; Santa Cruz), ERK1/2 (1:200; Santa Cruz), p-p38 (1:200; Santa Cruz), p38 (1: 100, Santa Cruz), p-JNK (1:300; Santa Cruz), JNK (1: 200; Santa Cruz) and β-actin (1:2000, Santa Cruz). The specific Jak2/Stat3 pathway inhibitor AG490 (25 μmol/ L, Calbiochem), ERK1/2 pathway inhibitor PD98059 (10 μM, Cell Signaling), Akt pathway inhibitors wort-mannin or LY294002 (10 nM, Calbiochem), and JNK pathway inhibitor SP600125 (5 μM, Calbiochem) were used to block the transcriptional signal in hUCMSCs. Membrane blocking, primary and secondary antibody incubations, and chemiluminescence reactions were conducted for each antibody individually according to the manufacturer's protocol. The intensity of each band was measured using a Kodak Digital Science 1D Image Analysis System (Eastman Kodak).
siRNA Preparation and Vector Construction
In siRNA preparation, two pairs of hairpin siRNA oligonucleotides for vimentin and Hsp27 were designed as previously described (8,10). The transfection protocols were performed in either FCS-cultured or hUCS-cultured hUCMSCs as previously described (33). Western blot analyses were performed to analyze protein expression from hUCMSCs in the two different serum cultures (FCS or hUCS) using specific antibodies to Hsp27 (1:200; Santa Cruz), vimentin (1:200; Chemicon), p65 antibody (1:100, Upstate Biotechnology), proliferative cellular nuclear antigen (PCNA, 1:100; Upstate Biotechnology), and β-actin (1:2000, Santa Cruz).
The Hsp27 overexpression vector, pCMV.SPORT6-hsp27, was constructed by inserting a 0.9 kb EcoRI/XhoI fragment of Hsp27 into the pCMV.SPORT6 vector (Invitrogen). As either hUCS- or FCS-cultured hUCMSCs grew to 60% confluence, cell transfection with 1 μg pCMV.SPORT6-hsp27 was conducted by electroporation as previously described (33).
In the luciferase assay, the cells were cotransfected using electroporation with 1 μg pGL2-vimentin-Luc (Invitrogen) and/or 1 μg pCMV.SPORT6-hsp27, and pBK-CMV LacZ (Invitrogen) as previously described (5).
Immunocytochemical Analysis and Senescence-Associated β-Gal Activity
For immunocytochemistry, cell cultures from hUCMSCs in the different serums (FCS or hUCS) were washed with PBS and fixed for 30 min at room temperature in 1% paraformaldehyde. After washing with PBS, the fixed cultured cells were treated for 30 min with blocking solution (10 g/L BSA, 0.03% Triton X-100, and 4% serum in PBS). Cells were incubated overnight at 4°C with an antibody against vimentin (1:200; Chemicon) conjugated with FITC (1:500, Jackson Immunoresearch), p65 conjugated with FITC (1:500, Upstate Biotechnology), and Hsp27 (1:300; Santa Cruz) conjugated with Cy3 (1:500, Jackson Immunoresearch). Finally, some of the preparation was lightly counterstained with DAPI, and then mounted. The preparations were analyzed with a Carl Zeiss LSM510 laser-scanning confocal microscope. Cells cultured in either FCS or hUCS were examined as previously described (6) for the senescence-associated biomarker β-Gal. In brief, hBMSCs were washed in PBS, fixed for 3–5 min (at room temperature) in 1% paraformaldehyde, washed, and incubated at 37°C (no CO2) with fresh senescence-associated β-Gal (SA β-Gal) stain solution (Cell Signaling Technology). Semiquantitative measurement of SA β-Gal staining was performed by calculating the percentage of SA β-Gal-positive cells.
Analysis of Telomerase Activity in hUCMSCs
A cell sample was prepared by plating passage 2 (p2) hUCMSCs at 100 cells/cm2 in a 10-cm diameter dish and incubating in FCS or hUCS for 7 days in order to quantitatively examine telomerase activity using a Telo-TAGGG PCR ELISA kit (Roche Molecular Biochemicals) according to the manufacturer's protocol as previously described (17).
Coimmunoprecipitation Analysis
Immunoprecipitation was as previously described (37). First, the cell lysate (300 μg) was incubated with protein A/G-agarose beads at 4°C for 6 h. Then, vimentin and Hsp27 antibodies were added and reacted for 6 h at 4°C. These immunocomplexes were incubated on protein A/G-agarose beads at 4°C overnight, and then examined by Western blot with anti-vimentin and anti-Hsp27 antibodies.
Electrophoretic Mobility Shift Assay (EMSA)
Detailed protocols to assess NF-κB DNA binding activity using EMSA have been described previously (27). The oligonucleotide probes corresponding to the NF-κB in the vimentin gene promoter and control Oct-1 were used (27). The oligonucleotides were nonradioisotope labeled using DIG Oligonucleotide 3′-End Labeling Kit (Roche). For supershift assays, 1 μg of anti-p65 antibody (1:100, Upstate Biotechnology) was added to the samples 1 h prior to the addition of labeled probes.
Statistical Analysis
All measurements in this study were performed blindly. The results were expressed as mean ± SEM. Oneway or two-way ANOVA with appropriate post hoc Newman-Keuls testing was used to evaluate mean differences among the control, and the treated groups. A value of p < 0.05 and p < 0.01 was taken as significant.
Results
Proliferative Efficacy of hUCMSCs in Different Culture Protocols
The cell yield from the low-density and high-density cultures treated with hUCS or hUCS+ was significantly higher than serum free or FCS (p < 0.05) (Fig. 1A–). Furthermore, calculations of the cumulative growth rate (cell counts) of hUCS-cultured hUCMSCs (from two different donors: No. 1 and No. 2) consistently yielded significantly higher cell counts than FCS-cultured hUCMSCs (p < 0.05) (Fig. 1D).

Cell density of hUCMSCs in media with different sera. (A) Representative pictures of the growth pattern of hUCMSCs incubated in four media with different serum content [serum free (SF), 10% FCS, 10% hUCS, and 10% hUCS] at the 3rd and 7th days, in phase microscopy. (B, C) Quantitative analysis of cellular yield of hUCMSCs from two different donors (green and blue) at 50 or 500 cells/cm2 incubated in the four types of serum. (D) Cumulative cell counts in cultures of hUCMSCs (from two different donors: No. 1 and No. 2) with hUCS or FCS plotted to identify the proliferative rate. Data are expressed as mean ± SEM. *p < 0.05 versus control. Scale bar: 50 μm.
Morphological Phenotypic Comparison Between hUCMSCs Grown in FCS and hUCS
Differences in morphology and immunophenotype between hUCMSCs incubated in FCS and hUCS were compared by visual inspection (Nikon, E600 microscope) and flow cytometry. hUCMSCs grown in hUCS were more homogenous and smaller than cells incubated in FCS (Fig. 2A, B). Cell size measurements indicated that FCS-cultured hUCMSCs were larger than hUCS-hUCMSCs (Fig. 2C). Flow cytometry of surface epitopes showed with mesenchymal phenotype grown in hUCS and FCS cultivation without any difference (Fig. 2D, E).

Morphological phenotypic comparison between hUCMSCs grown in FCS and hUCS. (A) Representative pictures of hUCMSCs grown in FCS or hUCS of different cellular density (50 and 500 cells/cm2) in phase microscopy. (B) hUCMSCs were more homogenous and smaller in hUCS than in FCS. (C) Quantitative analysis of cell size showed hUCMSCs cultured in FCS were larger than hUCMSCs cultured in hUCS. (D, E) Flow cytometric analysis for surface epitope of mesenchymal origin demonstrated similar results between cells grown in hUCS and FCS. Data are expressed as mean ± SEM. p < 0.05 versus control. Scale bars: 50 μm.
In Vitro Differentiation of hUCMSCs Growing in FCS or hUCS Into Neural Cells, Osteocytes, and Adipocytes
After 7–14 days of differentiation, half the cells in the dish exhibited refractile cell body morphologies with extended neurite-like structures arranged into a network. hUCMSC-derived neuroglial cells cultured in either FCS or hUCS were identified by immunostaining with specific antibodies against nestin, Tuj-1, NF, MAP-2, and GFAP, and then by expression of neural-specific genes (nestin, Tuj-1, MAP-2, and GFAP) using RT-PCR analysis (Fig. 3A). There were a higher percentage of neural progenitor cells in the hUCS-cultured hUCMSCs (nestin 45%, Tuj 52%, NF 50%, MAP-2 60%, and GFAP 40%) than in the FCS-cultured hUCMSCs (nestin 19%, Tuj 24%, NF 22%, MAP-2 37%, and GFAP 21%).
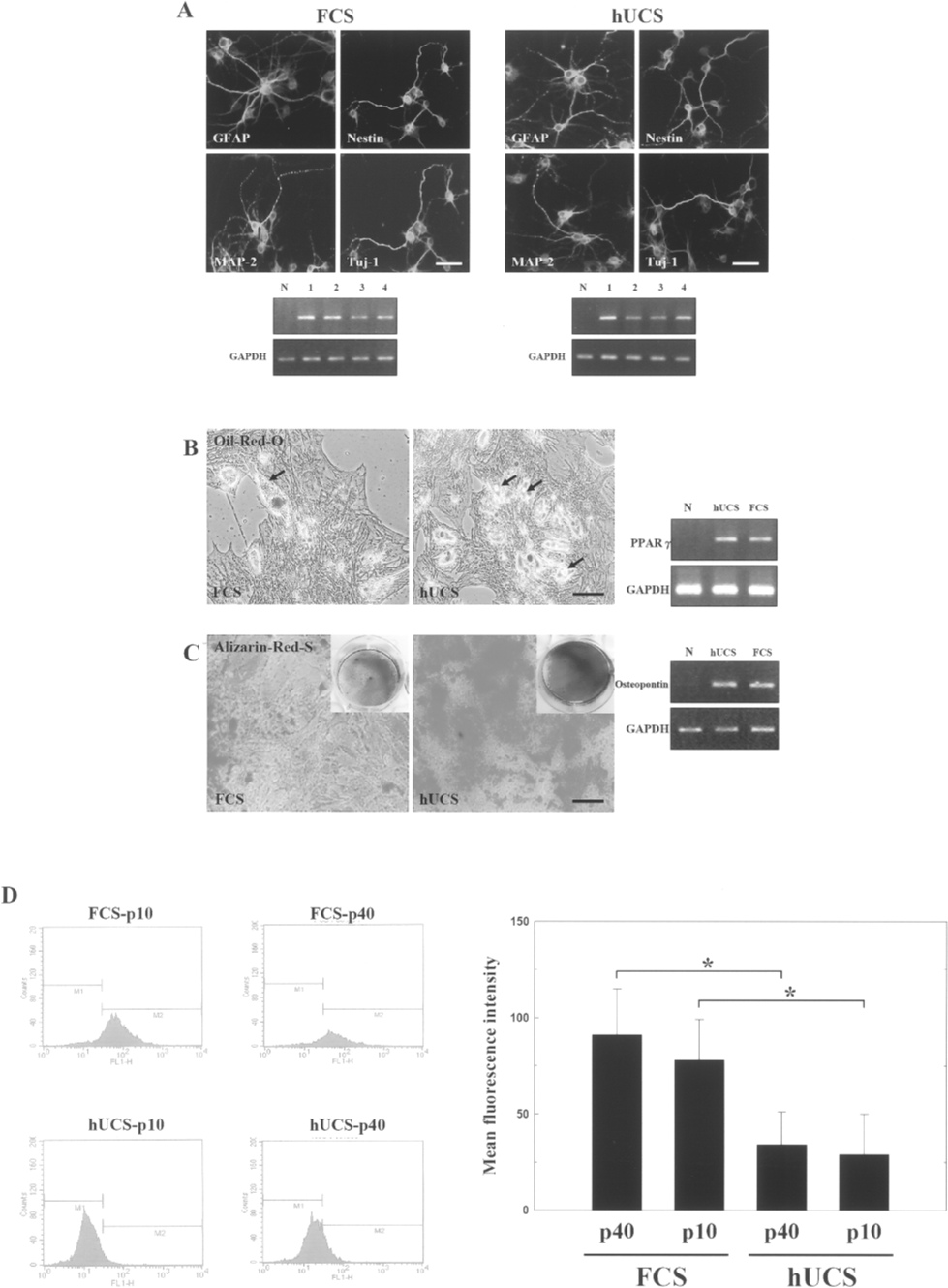
In vitro differentiation of hUCMSCs cultured in FCS or hUCS to neural cells, osteocytes, and adipocytes. (A) In neuroglial differentiation, morphologies of refractile cell bodies with extended neurite-like structures were arranged into a network, in phase microscopy. hUCMSC-derived neuroglial cells were identified by immunostaining against nestin, Tuj-1, MAP-2, and GFAP. RT-PCR analysis of untreated (lane N) and neurogenic formula-treated cells, specific genes (lane 1, GAPDH; lane 2, MAP-2; lane 3, nestin; and lane 4, Tuj-1) and GAPDH are shown. (B) Adipogenic differentiation shows morphological changes in the formation of neutral lipid vacuoles (black arrows), with almost all cells containing numerous Oil Red-O-positive lipid droplets. (C) Osteogenic differentiation shows numerous differentiated cells contained mineralized matrices, which were strongly stained by Alizarin Red-S. RT-PCR analysis of untreated (lane N), adipogenic and osteogenic formula-treated cells, with specific genes (PPAR-γ and osteopontin) and GAPDH (right panel). (D) In assessing the ROS formation, flow cytometry showed that p10 and p40 FCS-cultured hUCMSCs had significantly higher intracellular DCF fluorescence than p10 and p40 hUCS-cultured hUCMSCs. Data are expressed as mean ± SEM. *p < 0.05 versus control. Scale bars: 50 μm.
Adipogenic differentiation was apparent after 2 weeks of incubation with adipogenic medium supplement. Cell morphological changes showed formation of neutral lipid vacuoles, with almost all cells containing numerous Oil Red-O-positive lipid droplets (Fig. 3B). Similarly, on induction in an osteogenic medium, the treated cells cultured in either FCS or hUCS grew widely and contained mineralized matrices, which were strongly stained by Alizarin Red-S (Fig. 3C). In addition, RT-PCR analysis identified genes specific to PPAR-γ and osteopontin, showing that the cells had differentiated into adipocytes and osteocytes, respectively (Fig. 3B, C). In summary, hUCMSCs expanded in hUCS differentiated into adipocytes and osteocytes as readily as hUCMSCs expanded in FCS.
Generation of More ROS in FCS-Cultured Cells
The fluorescence intensity of DCF (the deesterified, oxidized product of DCF-DA) in the cells was determined by flow cytometry. FCS 10th (p10) and 40th (p40) passage FCS-cultured hUCMSCs had significantly more DCF intracellular fluorescence intensity (Fig. 3D) than hUCS-cultured hUCMSCs at p10 and p40.
Transplantation of hUCS-Cultured hUCMSCs Considerably Improves Neurological Deficit After Cerebral Ischemia
Behavioral measurement scores were all normalized to the baseline scores. From 14 to 28 days after transplantation, hUCS-cultured hUCMSC-treated rats (n = 10) exhibited significantly reduced body asymmetry in comparison to both FCS-cultured hUCMSC-treated (n = 10) and vehicle controls (n = 10) (Fig. 4A). Additionally, locomotor activity significantly increased between 14 and 28 days after cerebral ischemia in rats receiving hUCS-cultured hUCMSC treatment in comparison to FCS-cultured hUCMSC-treated and vehicle controls (Fig. 4B-D). Improvement in grip strength was greater in the hUCS-cultured hUCMSC-treated group than the FCS-cultured hUCMSC-treated and control groups (Fig. 4E).

hUCS-cultured hUCMSC implantation improves tissue plasticity in ischemic models. (A) Between 14 and 28 days after cerebral ischemia, hUCS-cultured hUCMSC-treated rats showed more recovery in the body swing examination than FCS-cultured hUCMSC-treated and controls. (B–D) Locomotor activity significantly increased between 14 and 28 days after cerebral ischemia in rats receiving hUCS-cultured hUCMSCs in comparison to FCS-cultured hUCMSC and vehicle controls. (E) hUCS-cultured hUCMSC-treated rats exhibited a better grip strength ratio than rats receiving FCS-cultured hUCMSC and controls. (F) Representative (coronal view) FDG-PET images and gross pictures of each ischemic rat's brain; there was a much bigger increase in metabolic activity in the right cortex (black arrow) of the hUCS-cultured hUCMSC-treated group than the FCS-cultured hUCMSC-treated group and the controls. (G) 3D confocal images of ischemic brain show many bis-benzimide labeled cells colocalized with the cellular marker MAP-2, GFAP, and Neu-N in both the hUCS- and FCS-cultured hUCMSC-treated rats' brain. (H) 3D images show hUCMSCs labeled with bis-benzimide colocalized with vascular phenotype vWF+ cells around the perivascular region of the ischemic mouse limb muscles. Quantitatively, CD31-immunoreactive blood vessel density revealed a larger increase in the limb muscles in the hUCS-cultured hUCMSC-treated group than in FCS-cultured hUCMSC-treated group and controls. (I) In blood perfusion analysis using LDPI, hUCS-cultured hUCMSC implantation led to significantly greater improvement of perfusion of ischemic limbs (black arrowhead) than FCS-cultured hUCMSC implantation and vehicle control. Data are expressed as mean ± SEM. *p < 0.05 and **p < 0.01 versus control.
Enhancement of Glucose Metabolic Activity in hUCS-Cultured hUCMSC-Treated Stroke Rats
MicroPET images showed a striking increase of FDG uptake over the right cortex of the hUCS-cultured hUCMSC-treated group (Fig. 4F) 1 week after each treatment. Semiquantitative measurement of relative glucose metabolic activity of the right hemisphere (relative to the nonstroke hemisphere) revealed significant enhancement in the hUCS-cultured hUCMSC-treated rats (n = 8) compared to FCS-cultured hUCMSC-treated rats (n = 8) and control rats (n = 8) (Fig. 4F).
Neurogenesis After Intracerebral Transplantation of hUCS-Cultured hUCMSCs Following Cerebral Ischemia
Some bis-benzimide-labeled cells colocalized with antibodies for GFAP, MAP-2, and Neu-N (Fig. 4G) in the penumbra of ischemic rat brains treated with FCS-or hUCS-cultured hUCMSCs. In quantitative analysis, the fractions of bis-benzimide-labeled cells colocalizing with specific markers MAP-2, GFAP, Neu-N, and vWF (~10%, ~13%, ~11%, and ~9%) in the hUCS-cultured cells, were significantly higher than with the FCS-cultured cells (~5%, ~6%, ~4%, and ~3%, respectively).
hUCS-Cultured hUCMSCs Increase Neovascularization and Improve Peripheral Blood Flow in Ischemic Limbs
Several cells labeled with bis-benzimide showed vascular phenotypes (vWF+ cells) around the perivascular regions (Fig. 4H) of the ischemic limb muscles of hUCS-cultured hUCMSCs-treated mice. Quantitative measurement of blood vessel density by CD31 immunoreactivity (Fig. 4H) showed that ischemic limbs from mice treated with hUCS-cultured hUCMSCs had more muscle neovascularization than groups with FCS-cultured hUCMSCs and controls (n = 6, each group). In evaluation of the blood perfusion improvement of ischemic limbs by LDPI, hUCS-cultured hUCMSCs treatment produced significantly greater recovery than FCS-cultured hUCMSCs and vehicle control mice (n = 8, each group; Fig. 4I).
Different Serum Incubation Induces Proteomic Expression Changes in hUCMSCs
A representative 2D gel image of protein lysates from hUCMSCs in FCS or hUCS is shown in Figure 5A and B. About 150 protein spots were resolved and identified with high reproducibility (>94%). Overall, around 12 protein spots were consistently up- or downregulated around twofold in triplicate experiments (data not shown). In particular, significantly increased expression of vimentin (point #12) and decreased expression of Hsp27 (point #4) were found in hUCMSCs cultured in FCS compared with hUCMSCs cultured in hUCS (data not shown).

Different proteomic expression in hUCMSCs grown in FCS or hUCS. (A, B) Representative 2D gel images of protein lysates from hUCMSCs cultured in FCS or hUCS. (C) Spot #12 and #4 in 2D gel. In Western blot analysis, significant upregulation of vimentin and downregulation of Hsp27 were found in FCS-cultured hUCMSCs compared with hUCS-cultured hUCMSCs. (D) Protein kinase expression assay showed activated Akt and activated JNK were upregulated in hUCS- and FCS-cultured hUCMSCs, respectively. Enhanced expression of Hsp27 and vimentin was inhibited by specific inhibitors LY294002 (LY*) and SP600125 (SP*) in hUCS- and FCS-cultured hUCMSCs, respectively. (E) Vimentin siRNA (Vim-si), transfected into FCS-cultured hUCMSCs, significantly downregulated the expression of vimentin compared to hUCS-cultured hUCMSCs. (F) In immunohistochemical analysis, vimentin fiber expression was more intense in FCS-cultured hUCMSCs (middle upper, and lower panel with magnification) than in hUCS-cultured hUCMSCs (left upper, and lower panel with magnification). The morphology of vimentin siRNA (Vim-si) transfected FCS-cultured hUCMSCs (right upper and lower panel with magnification) recovered to show a similar pattern to the hUCS-cultured hUCMSCs. (G) Representative pictures of SA β-Gal stain in FCS-cultured (p10 and p40) and hUCS-cultured hUCMSCs (p10 and p40). (H) Quantitative evaluation of percentage of SA β-Gal-positive cells on FCS-cultured, hUCS-cultured, and vimentin-siRNA transfected FCS (FCS+Vim-si)-cultured hUCMSCs. (I) Calculated telomerase activity (PCR-ELISA) in cultures of hUCMSCs were plotted to identify the telomerase activity in the hUCS-cultured, FCS-cultured, GFP-siRNA transfected FCS (FCS+GFP-si)-cultured, and FCS+Vim-si-cultured hUCMSCs. (J) In a double immunofluorescence study, distinct distributions of Hsp27 (cytoplasmic) and vimentin (perinuclear) colocalized in both hUCS- and FCS-cultured hUCMSCs. (K) Co-IP analysis proved that Hsp27 interacts with vimentin. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01 versus control. Scale bars: 50 μm.
Different Expression of Vimentin and Hsp27 in FCS- and hUCS-Cultured hUCMSCs
Western blot analyses revealed a significant increase in vimentin expression in the FCS-cultured hUCMSCs (Fig. 5C). In contrast, there was significantly more Hsp27 in the hUCS-cultured hUCMSCs than in the FCS-cultured hUCMSCs (Fig. 5C). The level of activated Akt in the hUCS-cultured hUCMSCs was significantly higher than in FCS-cultured hUCMSCs, while there was significantly more activated JNK in the FCS-cultured hUCMSCs (Fig. 5D). The upregulation of Hsp27 and vimentin was blocked by specific inhibitors LY294002 and SP600125 in hUCS- and FCS-cultured hUCMSCs, respectively (Fig. 5D).
In addition, vimentin expression was significantly downregulated by transfecting vimentin-siRNA into FCS-cultured hUCMSCs (p25) (Fig. 5E). hUCMSCs cultured in hUCS showed characteristic small cell shapes and shorter and thinner vimentin filaments, while hUCMSCs cultured in FCS exhibited a dense vimentin network with numerous long, thick vimentin filaments (Fig. 5F). However, FCS-cultured hUCMSCs transfected with vimentin-siRNA had small, short cell shapes similar to hUCS-cultured hUCMSCs (Fig. 5F).
Increased SA β-Gal Activity in FCS-Cultured hUCMSCs
In β-Gal staining, senescent hUCMSCs expressed β-Gal that was detected in single cells by X-Gal, which forms a local blue precipitate (Fig. 5G). Quantitatively, there was a significantly greater increase in the percentage of SA β-Gal-positive cells in hUCMSCs cultured in FCS (p10 and p40) than in hUCMSCs cultured in hUCS (p10 and p40) (Fig. 5H). However, the percentage of SA β-Gal-positive cells in FCS-cultured hUCMSCs transfected with vimentin-siRNA was similar to that for hUCS-cultured hUCMSCs (Fig. 5H).
Telomerase Activity of hUCS-Cultured hUCMSCs Is Greater Than That of FCS-Cultured hUCMSCs
Telomerase activity was significantly higher in the hUCS-cultured hUCMSCs than in the FCS-cultured hUCMSCs (Fig. 5I). In addition, telomerase activity in the FCS-cultured hUCMSCs transfected with vimentin-siRNA was significantly increased in comparison to FCS-cultured hUCMSCs transfected with GFP-siRNA (Fig. 5I).
In IHC of both hUCS-cultured hUCMSCs and FCS-cultured hUCMSCs, vimentin was distributed in cytoplasmic aggregates concentrated in the perinuclear caps, and arranged along tracks which seemed to emanate from the nuclear periphery (Fig. 5J). Granular Hsp27 immunostaining was seen predominantly in the cytoplasm, and along tracks coinciding with vimentin filaments (Fig. 5J). Merging of the IHC images of Hsp27 and vimentin showed that they coincided in both the cytosol and perinuclear regions (Fig. 5J).
To verify whether Hsp27 binds to vimentin, we conducted Co-IP analysis with specific antibodies for Hsp27 and vimentin (Fig. 5K). Results indicated that vimentin exists in a complex with Hsp27 in the intact hUCMSCs.
Overexpression of Hsp27 Downregulates Expression of Vimentin Through Inhibition of NF-κB Signaling Pathway
Western blot revealed significantly decreased expression of vimentin in the FCS-cultured hUCMSCs transfected with pCMV.SPORT6-hsp27 compared to those with vector only (Fig. 6A), indicating that induction of Hsp27 overexpression downregulates the cellular expression of vimentin.

Overexpression of Hsp27 reduced expression of vimentin by blocking the nuclear translocation of p65/NF-κB. (A) In either FCS- or hUCS-cultured hUCMSCs transfected with pCMV.SPORT6-hsp27, the expression of vimentin and the nuclear level of p65 were significantly decreased in the hUCMSCs after Hsp 27 overexpression compared to that with vector only. (B) In immunofluorescent analysis of subcellular localization of p65/NF-κB using confocal microscopy in either FCS- or hUCS-cultured hUCMSCs, the cells were fixed and stained with PI (nucleus marker, red) and anti-p65 (FITC-conjugated, green). (C) In EMSA assays, the binding activity of NF-κB (indicated by an asterisk *) was evaluated with or without induced Hsp 27 overexpression in both FCS- and hUCS-cultured hUCMSCs. (D) After cotransfection experiment with pCMV.SPORT6-hsp27 and pGL2-vimentin-Luc or pBK-CMV LacZ, relative luciferase activity was determined with a luciferase assay system and was normalized to LacZ expression. Data are expressed as mean ± SEM. *p < 0.05 versus control. Scale bar: 50 μm.
Nuclear p65 levels in FCS-cultured hUCMSCs were reduced after Hsp27 overexpression in comparison to the levels in the cytosol (Fig. 6A), suggesting that induction of Hsp27 stimulates p65 translocation from the nucleus to cytosol. Similar inhibition of p65 translocation from cytosol to nucleus by Hsp27 overexpression was further demonstrated by immunofluorescent staining of p65 using confocal microscopy in the FCS-cultured hUCMSCs (Fig. 6B).
EMSA was used to analyze the functional status of NF-κB protein complexes induced by Hsp 27 overexpression. Binding of nuclear extract to the NF-κB element was decreased by Hsp27 overexpression in both FCS- and hUCS-cultured hUCMSCs (Fig. 6C). Hsp27 overexpression had no effect on binding to the Oct-1 element (Fig. 6C). To confirm transcriptional deactivation of the NF-κB element in vimentin by Hsp27 overexpression, we cotransfected a reporter construct containing the NF-κB element found in the regulatory region of the vimentin gene and pCMV.SPORT6-hsp27 plasmids. Luciferase activity was significantly reduced after cotransfection with pCMV.SPORT6-hsp27 and NF-κB-containing reporter plasmids in hUCMSCs compared with cells without Hsp 27 overexpression (Fig. 6D).
Discussion
Human mesenchymal stem cells (hMSCs) from bone marrow and umbilical cord can differentiate into mesenchymal cells such as osteoblasts, adipocytes, chondrocytes, hematopoietic-supporting stroma, and neural cells both ex vivo and in vivo (13,30) and the efficacy of administering hMSCs for the treatment of various diseases has been demonstrated in human clinical trials (16,23). However, these trials have all used hMSCs cultured in FCS according to the Institutional Review Board approved protocol (16,23). FCS cultivation may enhance cellular DNA damage due to free radical formation, and immunological rejection of the transplanted cells (11). In other words, FCS culture conditions expose hMSCs to considerable oxidative stress. Oxidative stress causes genome and proteome damage, and promotes senescence and aging (14). The biological relevance of oxidative stress in the survival and self-proliferation capacity of stem cells has been demonstrated in stem cells with a defective cell cycle checkpoint activator (18). Finally, the insult caused by oxidative stress on the implanted stem cells might be one of the major causes of poor cellular engraftment in one clinical trial patient (16).
Here, we demonstrated that hUCS culture of hUCMSCs induced less ROS formation than FCS culture. In addition, hUCS-cultured hUCMSCs showed greater self-proliferative capacity and less cellular senescence than FCS-cultured hUCMSCs. Applying FCS-cultured stem cells in clinical trials has raised potential hazards that cannot be neglected; however, up to now there has been no definite strategy to resolve these problems. In recent reports, some improved protocols have been developed, including serum-free medium (29), selenium supplement (9), and human plasma supplement (28), but these protocols have not yet been approved. Because it is necessary to add serum at a concentration of over 10% to the culture medium, application of autologous adult human serum is not practical in the clinic (38). In addition, in our study, we have demonstrated that hUCS-cultured hUCMSC implantation could significantly enhance tissue plasticity in cerebral and hindlimb ischemic models compared to FCS-cultured hUCMSCs. In conclusion, we have developed a feasible stem cell culturing strategy using hUCS for future clinical application.
The interaction of Hsp27 with vimentin seems to cooperate in many conditions, including protecting cells from stress-induced aggregation in vitro (35), maintaining cytoskeletal integrity and promoting cell survival in vivo (35). However, few reports have investigated the correlation between Hsp27, vimentin, and cellular senescence. Some recent studies suggested that oxidative stress and inflammation inducing cellular senescence might upregulate the vimentin expression through activation of NF-κB (32). Because NF-κB is located on the regulatory element of the human vimentin promoter (27), Perkins et al. demonstrated that cellular senescence might be due to NF-κB-activation induced gene transcription of vimentin (34). On the other hand, overexpression of Hsp27 might block the nuclear translocation of p65 in intestinal epithelial cells, and then suppress the activation of NF-κB (20). Here we have demonstrated that overexpression of Hsp27 downregulated the expression of vimentin by inhibiting the nuclear translocation of p65 and reducing NF-κB-DNA binding in the FCS-cultured hUCMSCs. Therefore, we conclude that: 1) different serum conditions (i.e., cells cultured in different environments) can modulate the expression of Hsp27 and vimentin through different signaling pathways, and 2) biological interaction between Hsp27 and vimentin are two of the specific factors that determine the fate of cellular senescence. Interaction between Hsp27 and vimentin may modulate the degree of senescence in hUCS- and FCS-cultured hUCMSCs. We conclude that differential regulation of the expression of vimentin and Hsp 27 plays an important role in cellular senescence.
Stem cell transplantation might be one of the better strategies to treat various neurodegenerative disorders (2,42). However, limited efficacy of stem cell transplantation is due to the fact that large numbers of transplanted stem cells cannot survive to engraft for a long time (31). The reason of poor stem cell engraftment into host brain is likely to be affected by the ischemic environment and cellular senescence after transplantation, which is lack of growth factors, and the loss of survival signals from tissue matrix and cell to cell interactions (37). In previous study, there are some strategic interventions to overcome these problems to enhance cell survival immediately after transplantation, including the delivery of neuroprotective factors produced by the stem cells (12), and the replacement of other nonneuronal cells (25). Regarding this study, genetically modification to induce Hsp27 protein production in the hUCMSCs might be a good method to promote the neurological recovery after stroke. Although the treatment approach listed above seemed to be very exciting, it is still in preliminary stage and clinically unfeasible at the current time. We need to find simple and applicable strategies in the stem cell treatment for the human stroke. Therefore, hUCS culturing protocol for human stem cell implantation might be a safe and effective method for clinical application.
Footnotes
Acknowledgments
We thank Dr. H. Wilson and Ms. M. Loney for their critical reading of this manuscript. This work was supported by grants from Academia Sinica (AS92IMB3 to H.L.) and the National Science Council (NSC96-2314-B-303-002 to H.L., Taiwan).