
Abstract
Human fetal retinal cells have been widely advocated for the development of cellular replacement therapies in patients with retinal dystrophies and age-related macular degeneration. A major limitation, however, is the lack of an abundant and renewable source of cells to meet therapeutic demand, although theoretically this may be addressed through the use of immortalized retinal progenitor cell lines. Here, we have used the temperature-sensitive tsA58 simian virus SV40 T antigen to conditionally immortalize human retinal progenitor cells isolated from retinal tissue at 10–12 weeks of gestation. We show that immortalized human fetal retinal cells retain their progenitor cell properties over many passages, and are comparable with nonimmortalized human fetal retinal cultures from the same gestational period with regard to expression of certain retinal genes. To evaluate the capacity of these cells to integrate into the diseased retina and to screen for potential tumorigenicity, cells were grafted into neonatal hooded Lister rats and RCS dystrophic rats. Both cell lines exhibited scarce integration into the host retina and failed to express markers of mature differentiated retinal cells. Moreover, although immortalized cells showed a greater propensity to survive, the cell lines demonstrated poor long-term survival. All grafts were infiltrated with host macrophage/microglial cells throughout their duration of survival. This study demonstrates that immortalized human fetal retinal progenitor cells retain their progenitor characteristics and may therefore have therapeutic potential in strategies that demand a renewable and consistent supply of donor cells for the treatment of degenerative retinal diseases.
Introduction
Retinal degenerative diseases, such as inherited retinal dystrophies and age-related macular degeneration (AMD), are a major cause of visual impairment and blindness in the industrialized world and represent a considerable economic and social burden to society. These diseases have proven to be largely refractory to treatment despite enormous efforts to develop effective therapeutic strategies. Currently available treatments, including the antiangiogenics, at best slow disease progression in a minority of patients while none are able to halt the disease or restore lost photoreceptors. Recent preliminary evidence suggests that gene therapy may prove efficacious in monogenic inherited retinopathies (3,22), although this approach is less likely to be effective in genetically dominant retinal dystrophies or where photoreceptors have already been lost. Cell-replacement therapies, which have the potential to resolve this latter obstacle, have consequently attracted considerable attention. In particular, the utility of stem and progenitor cells to reconstitute function in retinal disease is increasingly being investigated.
The choice of cell origin has evoked substantial debate regarding the relative merits of embryonic stem (ES) cells versus tissue-derived stem/progenitor cells, with both having been investigated for their capacity to differentiate into retinal precursors and integrate into the retina (12,15,16,21,26). The use of tissue-derived stem/progenitor cells, however, is particularly tempting as they are already partially committed and retain their ability to generate different cell types up to the final cell division (23). Nevertheless, there is restricted availability of retinal progenitor cells, especially from fetal tissue, with a limited replicative potential in vitro, reducing their utility (2).
To overcome the limitations of donor cell supply, immortalization techniques have previously been used to produce large numbers of cells including immortal human retinal pigment epithelial cells for the potential treatment of AMD (7,20) and brain-derived stem cells for the treatment of a variety of CNS diseases (10,11,24,28,29). There are several additional advantages of generating an immortalized cell line for cell therapy. Firstly, despite the potential hazards of immortalization, an immortalized stem/progenitor cell line can conform more readily to various commercial and safety requirements with regard to therapeutic application. And secondly, such lines are comprised of a homogenous population that is more tractable to further genetic manipulation should this improve their efficacy.
To address the issue of supply of human fetal retinal progenitor cells as a possible source of cells for the treatment of retinal degeneration, we have conducted a proof-of-concept study to evaluate the effect of immortalization on cell phenotype. To this end we have generated a temperature-sensitive simian virus (SV) 40 T immortalized cell line derived from a 10–12-week-old human retina. We demonstrate that these cells retain progenitor-like characteristics but in their current configuration do not integrate or survive for extended periods in an animal model of retinal degeneration.
Materials and Methods
Reagents
All reagents were purchased from Sigma unless specified otherwise. Antibodies to the following epitopes were used: neurofilament 160 kDa [NF-160, mouse monoclonal antibody (mAb) 1:1000, Chemicon], glial fibrillary acidic protein (GFAP, mouse mAb 1:500), cytokeratin 8 (mouse mAb 1:2000, Chemicon), anti-human nuclear antigen (HNA, mouse mAb 1:1000, Chemicon), anti-human mitochondria (MIT, mouse mAb 1:2000, Chemicon), anti-CD68 (mouse mAb 1:1000 Serotec), nestin [goat polyclonal antibody (pAb) 1:200, Santa Cruz], S-opsin (goat pAb 1:500, Santa Cruz), neurofilament 200 kDa (NF-200, rabbit pAb 1:5000, Chemicon), rhodopsin (rabbit pAb 1:200, Santa Cruz), Ki67 (rabbit pAb 1:2000, Novocastra Laboratories), recoverin (rabbit pAb 1:1000, Chemicon), and calretinin (rabbit pAb 1: 5000, Swant).
Human Fetal Retinal Cell Isolation and Culture
The human retinal cells used in this study were derived from 10–12-week gestational fetal retinal tissue following termination in accordance with national (UK and US), legal, and ethical guidelines. The GuRt09 immortalized cell line was derived from tissue obtained from the MRC Tissue Bank (Hammersmith Hospital, London, UK); and the GS076 nonimmortalized cell line was derived from tissue obtained from Advance Bioscience Resources (Almeda, CA, USA). The protocol of this study adhered to the tenets of the Declaration for Helsinki regarding research involving human tissue.
Tissue was collected in ice-cold HBSS without Ca2+/Mg2+ (Gibco), containing 1 mM N-acetyl cysteine. The neural retina was removed from the eyes and cleaned of debris under a dissection microscope. The tissue was dissociated by first dicing with a scalpel followed by incubating in 0.25% w/v trypsin (Cambrex Biosciences, Belgium) in DMEM/F12 (Gibco) containing 0.025 U/ml Benzonase® (Merck) for 15 min at 37°C. Following dissociation, twice the volume of trypsin inhibitor solution (5.5 mg/ml soybean trypsin inhibitor dissolved in DMEM/F12, containing 1% human serum albumin, Grifols, and 0.025 U/ml Benzonase®) was added. Cells were centrifuged at 800 x g for 5 min, then resuspended in serum-free human culture media as previously described (28) with the addition of l-thyroxine (T4; 400 ng/ml); tri-iodo-thyronine (T3; 337 ng/ml), and heparin sodium salt (10 U/ml), defined as human medium. The yield of cells isolated from the neural retina of two eyes was approximately 3 × 106, with 90% viability as judged by trypan blue exclusion.
From each preparation isolated cells were plated out in human culture media into two laminin-coated wells (coated at 5 μg/ml in DMEM/F12 for 1 h at 37°C followed by one wash with DMEM/F12) of a six-well plate and maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. Upon reaching confluency cells were passaged into 1–5 -g/cm2 fibronectin-coated flasks (Invitrogen). Human fetal retinal progenitor cells harvested from individual donors differed in their ability to attach and grow as monolayers (data not shown). One representative culture was selected (designated GS076). This nonimmortalized cell line was cultured at 37°C in the same culture medium used for the immortalized cells (human medium) with the addition of EGF, bFGF, and 5% fetal bovine serum (FBS). This cell line was cultured for 10 passages over which period a comparison was made with the immortalized human fetal cell line described below (passage 8 and 22).
Generation of an Immortalized Human Fetal Retinal Cell Line (GuRt09)
Fetal retinal cell cultures at passage two were infected at 50% confluency for 8 h with fresh human media containing a retrovirus carrying the temperature-sensitive TsT-SV40 antigen gene (pLNCX-SV40-U19tsA58) in the presence of 4 μg/ml polybrene. Three successive 8-h infections were carried out followed by antibiotic selection (150 μg/ml Geneticin®, Gibco). To generate clonal lines, surviving cells were plated at a density of 20–40 cells/cm2 onto laminin-coated petri dishes and following 2–3 weeks of growth, cell colonies were harvested using 8 × 8-mm cloning rings. A total of 11 clones were isolated, expanded, and frozen down in 10% DMSO in human medium in the presence of bFGF and EGF. One clone, designated GuRt09, was selected for this study based on its long-term stability over several passages.
The GuRt09 immortalized human fetal retinal cell line was cultured at the permissive temperature of 33°C in human medium supplemented with 10 ng/ml bFGF (Peprotech) and 20 ng/ml EGF (Peprotech). Cell medium was replaced twice weekly and cells seeded at a density of approximately 10,000 cells/cm2 at 33°C or at the nonpermissive temperature of 37°C to facilitate cell differentiation. The GuRt09 immortalized cell line was expanded, with no differences being observed in morphology or expression profile, up to passage 32 with all analysis being conducted on cells between passage 8 and 22.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
mRNA was extracted from both the GS076 nonimmortalized cell line and the GuRt09 immortalized cell line using TRIzol reagent (Invitrogen, UK), according to the manufacturer's instructions. The resulting RNA preparations were quantified using a spectrophotometer and subsequently DNase treated (Promega, Madison, WI, USA). Single-stranded cDNA was prepared with 4 μg RNA using a Moloney Murine Leukemia virus reverse transcriptase kit (Promega). Alongside the synthesis of cDNA, controls lacking reverse transcriptase were also run simultaneously, referred to as “no RT” controls. The expression of a number of key retinal progenitor and neural-retinal cell type-specific genes (Table 1) in these cell lines was then analyzed by RT-PCR. The cDNA templates were run in standard PCRs using Go Taq Flexi Kit (Promega), with amplification conditions as follows: 95°C for 5 min followed by 35–40 cycles of 94°C for 1 min, annealing temperature appropriate to primer pair (Table 1) for 1 min, and 72°C extension for 1 min, followed by a final extension at 72°C for 4 min. PCRs of human fetal retinal cell cDNA were carried out alongside both water (no template control, NTC) as well as no reverse transcriptase (no RT) controls. Prior to use on human fetal cell cDNA, the specificity of all primers were tested on a phage (λ GT10) human retinal cDNA library (generously supplied by Dr A. Hardcastle, UCL Institute of Ophthalmology).
Primer Sequences Used for RT-PCR of Retinal-Associated Markers

GenBank accession number and appropriate reference for primer pairs (where not designed in-house) are shown.
Immunocytochemistry
Cells were cultured in 35-mm petri dishes at a density of 10,000 cells/cm2 and grown for 1 week under various conditions. Cells were fixed with 4% paraformaldehyde (PFA) for 30 min at 4°C and following several washes with 0.1 M phosphate buffered saline (PBS, pH 7.4) were permeabilized with 0.3% Triton X-100 in 0.1 M PBS (PBS-TX) for 2 min at room temperature. After permeabilization, cells were washed and blocked in 5% normal donkey serum (NDS) (Jackson ImmunoResearch) for 30 min at room temperature. Samples were then incubated with primary antibodies (see above) in the presence of 1% NDS at 4°C overnight and following three successive washes incubated with appropriate fluorophore-labeled secondary preabsorbed antibodies (Jackson ImmunoResearch) at 1:200 in 2% NDS for 30 min. Labeled cells were mounted in Vectashield mounting media containing 4′6-diamindino-2-phenylindole dihydrochloride (DAPI; Vector Laboratories, Peterborough, UK) and selected random fields imaged by epifluorescent and confocal microscopy. A minimum of three independent fields were then used to quantify the percentage of positively stained cells.
Transplantation Into Neonates and RCS Dystrophic Rats
The nonimmortalized GS076 and the immortalized GuRt09 human fetal retinal cell lines were transplanted into neonatal [postnatal day 3 (P3)] normal hooded Lister rats (n = 20), adult RCS dystrophic (rdy-/p+) rats (n = 44) or RCS congenic (nondystrophic) controls (n = 8). Cells were transplanted into dystrophic rats during the active phase of retinal degeneration (4–8 weeks of age). All host animals were maintained in a 12-h light/dark cycle with food and water ad libitum. All procedures were performed in accordance with UK Home Office regulations under the Animals (Scientific procedures) Act 1986.
Two days prior to grafting the human cell lines, adult RCS rats were given oral cyclosporine A (Sandoz, 210 mg/L of drinking water) ad libitum to suppress the host immune system. Immunosuppression of neonatal hosts was achieved by addition of cyclosporine to the dams' drinking water when pups were 1 day old, 2 days prior to surgery at P3. For the transplantation of cells, animals were anesthetized by intraperitoneal injection using a mixture of ketamine (50 mg/kg) and xylazine (10 mg/kg). Cell suspensions of GS076 or GuRt09 were prepared in DMEM and stored on ice prior to the transplantation procedure. Adult RCS rats were placed under an Olympus operating microscope and a cell suspension (2 μl at 5 × 104 cells/μl in DMEM) was injected through the sclera into the subretinal space using a fine glass pipette (internal diameter 200 μm) attached to a 10-μl Hamilton syringe. For neonatal hosts, a 1-μl injection (5 × 104 cells) was made into the vitreous cavity via a small opening in the eyelids (made using microsurgical scissors). After surgery anesthesia was counteracted with Antisedan® (atipamezole hydrochloride 5 mg/kg).
Following transplantation surgery, all host animals were maintained on the same dose of cyclosporine for the duration of the experiment.
Tissue Preparation and Immunohistochemical Labeling
The survival of nonimmortalized and immortalized cells following subretinal grafting in vivo was evaluated in animals following 5, 26, and 63 days posttransplantation. Rats were deeply anesthetized with sodium pentobarbital (200 mg/kg) and perfused with PBS, followed by 4% paraformaldehyde (PFA). The eyes were removed and further postfixed overnight at 4°C. After washing in PBS, the lens was removed and the eye cryoprotected in 30% sucrose before rapid freezing in OCT (Tissue Tek®). Cryostat sections (14 μm) were thaw-mounted onto charged slides (BDH).
Grafted cells were identified by immunofluorescent staining for a combination of anti-human nuclear antigen and anti-human mitochondrial antigens (referred to as HNA-MIT). However, our previous unpublished observations indicate that subpopulations of macrophages/microglia can be autofluorescent, a phenomenon that may lead to false-positive identification of grafted cells. In order to control for this, and to unambiguously identify transplanted human cells, we developed an histological protocol to visualize these cells concurrently with host macrophage/microglia. This technique employs peroxidase staining with the ED1 antibody (specific for CD68 on rat macrophages and microglia) prior to labeling other relevant antibodies with fluorescence. After blocking with 5% normal horse serum (NHS) in PBS with 0.3% Triton X-100 (PBS-TX), sections were incubated in mouse anti-CD68 antibody in PBS-TX with 1% NHS overnight at room temperature. After washing, sections were incubated for 2 h in PBS-TX containing biotinylated horse anti-mouse secondary antibody (Vector, 1: 200), washed again, and then incubated for 1 h in a 1:50 dilution of Avidin-Biotin-Peroxidase Complex (Vector, ABC Elite kit) in PBS-TX. Following washing, sections were reacted in a 0.05% solution of 3,3′-diaminobenzidine (DAB) with nickel and cobalt chloride (1) and 0.03% hydrogen peroxide for 30 s. The peroxidase reaction was terminated by extensive washing in PBS. This provides a bright-field signal for macrophage/microglia that does not interfere with fluorescent antibody labeling with other epitopes. Slides were then blocked with 5% normal donkey serum (NDS, Jackson ImmunoResearch) in PBS for 1 h followed by overnight application of the relevant combination of primary antibodies (see Immunocytochemistry section above) diluted in PBS with 1% NDS. After washing, appropriate combinations of fluorescent secondary antibodies were applied in PBS plus 2% NDS. After washing, cell nuclei were counterstained with DAPI, washed, and coverslipped with Vectashield (Vector Labs).
Confocal Microscopy
Fluorescently stained cells were examined and images captured using a Zeiss LSM 510 confocal laser scanning microscope with LSM software. Unless otherwise indicated, figure images are presented as Z stack projections.
Results
Characterization of Human Fetal Retina at 12 Weeks of Gestation
As progenitor cell lines were to be derived from human fetal retina at 10–12 weeks of gestation, we first investigated human donor retinae taken from the beginning of the second trimester of development (week 12). Immunohistochemical analysis revealed a nestin-positive neural retina segregated into two distinct anatomical regions. The inner retina was characterized by calretinin-expressing retinal ganglion cells, the axons of which could be seen exiting the eye to form the optic nerve (Fig. 1A). At this stage, inner retinal neurons did not express neurofilament proteins, which have been reported to appear at around week 20 of development (31). The outer retina was composed of a neuroblastic zone containing cells in active phases of the cell cycle (Ki67 positive) (Fig. 1B). Recoverin-positive outer retinal progenitor cells were clearly visible in this tissue (Fig. 1C, D) and were intimately associated with the retinal pigment epithelium (RPE), which was fully pigmented at this stage (Fig. 1C). No immunoreactivity was evident for differentiated retinal ganglion/horizontal cells (neurofilaments) or photoreceptors (rhodopsin and S-opsin) at this stage of development, nor were there any obvious signs of central–peripheral gradients of differentiation observed with any of markers used. The results of immunohistochemical staining on the retinal tissue are summarized in Table 2.

Human fetal retina at the start of the second trimester (12–13 weeks). Ganglion cells of the developing inner retina express calretinin and their axons form the optic nerve (A). (B) Nestin staining in red with Ki67-positive (green) neural retinal progenitor cells actively engaged in the cell cycle. Recoverin-positive cells were found in close association with the RPE (dashed boxes in C and D). The photomicrographs in (C) and (D) show different channels from the same field of view. Antigens of interest are color coded; DAPI-stained nuclei are blue. Optic nerve (ON); retinal pigment epithelium (RPE); neuroblastic zone (NBZ). Scale bars: (A–C) 50 μm; (D) 10 μm.
A Summary of Immunostaining Results for Nonimmortalized and Immortalized Human Fetal Retinal Cells In Vitro and In Vivo

Positive (+) or negative (-) immunostaining in vitro or in tissue sections (12-week human retina or grafted cells in vivo). Following transplantation of cells into the rat eye, nestin expression was found in nonimmortalized (NI) cells, while Ki67 staining was restricted to immortalized (I) cells.
GS076 and GuRt09 Cell Lines Express a Similar Pattern of Progenitor and Cell-Specific Markers
At the nonpermissive temperature of 37°C, both the nonimmortalized GS076 and the immortalized GuRt09 cell lines grew as monolayers (Fig. 2A, B) and expressed mRNA for the neural retinal progenitor cell markers Hes1, Sox2, and nestin (Fig. 2, right side). However, they also expressed genes for retinal ganglion/horizontal cells (neurofilaments, βIII tubulin) and cone photoreceptors (recoverin and S-opsin), which are characteristic of a more differentiated cell phenotype. To establish whether this translated into protein expression and to determine the extent of expression of these and other cell type-defining markers we performed immunocytochemical analyses. Expression patterns were also quantified by enumerating the number of positively stained cells. The nonimmortalized GS076 cells had fewer positively stained cells for the proliferation marker Ki67 (Fig. 3A) with 20% expressing this marker compared to 82% in the GuRt09 cell line (Fig. 3C). The immortalized cells exhibited similar Ki67 staining at both the permissive (33°C) and nonpermissive (37°C) temperatures (data not shown), suggesting either that the immortalizing gene is transcriptionally leaky and/or that these cells have a robust underlying proliferative capacity.

Phase contrast images of immortalized human fetal retinal cells (A) and nonimmortalized human fetal retinal cells (B). Influence of immortalization on the gene expression profile of cultured human fetal retinal cells (C). RT-PCR shows that markers of early neural retinal progenitor cells, retinal ganglion cells, and photoreceptors can be found in both nonimmortalized (GS076) and immortalized (GuRt09) cultures. As controls, samples were run concurrently that lacked reverse transcriptase (no RT). The no template control (NTC) is shown to the right. Scale bars: (A) 60 μm; (B) 70 μm.
The GS076 and GuRt09 cultures both contained cells positive for nestin (92% and 88%, respectively), neurofilament 160 (NF-160) (79% and 86%, respectively), neurofilament 200 (NF-200) (39% and 62%, respectively), and recoverin (75% and 87%, respectively), while only the nonimmortalized culture showed cells positive for S-opsin (5%), as well as occasional cytokeratin 8-positive cells (2%), possibly representing RPE cells (Fig. 3, Table 2). More diversity in the cell phenotypes present would be expected in the GS076 culture as it is a polyclonal-derived cell line, as opposed to the GuRt09 line, which was derived from a single clonal cell.
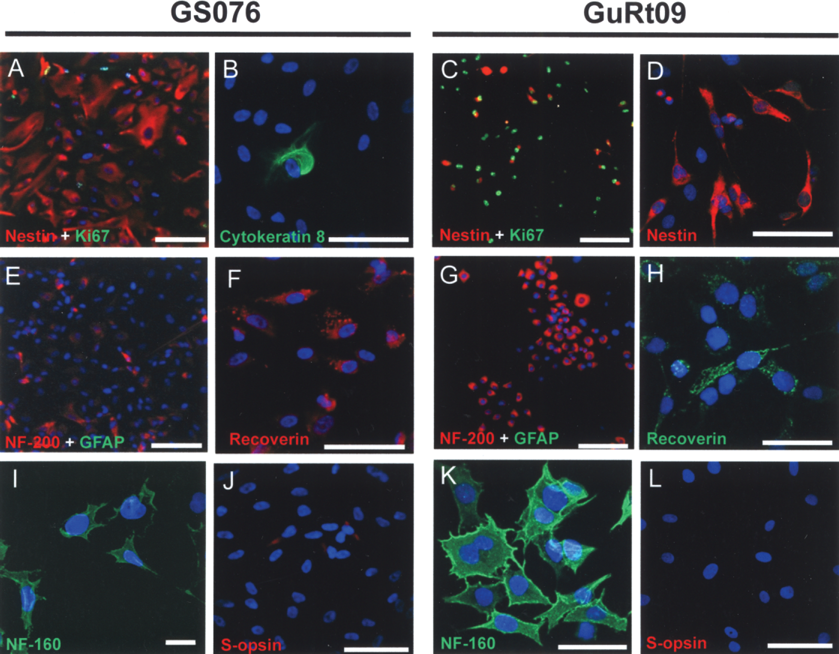
Nonimmortalized (GS076) (left panels) and immortalized (GuRt09) (right panels) human fetal retinal cells could be expanded as a monolayer while retaining markers of early neural retinal cell types. The majority of nonimmortalized cells at passage 10 expressed nestin (A). Occasional cells were found to be positive for the RPE marker cytokeratin 8 (B). The majority of immortalized cells expanded at 33°C at passage 14 expressed nestin and Ki67 (C, D). Neurofilament staining provides evidence for acquisition of a neural retinal phenotype (E, G, I, K), while recoverin expression indicates the emergence of photoreceptor/bipolar cell phenotypes (F, H) in both cell types. A subset of nonimmortalized cells was found to be positive for S-opsin (J) while this marker was not detected in the immortalized cells at the translational level (L). Antigens of interest are color coded; DAPI-stained nuclei are blue. Scale bars: (A, C, E, G) 100 μm; (B, D, F, I, J, L) 50 μm; (H, K) 30 μm; (I) 10 μm.
Both nonimmortalized (GS076) and immortalized (GuRt09) human fetal retinal cells were able to form neurospheres when plated on to uncoated tissue culture plastic ware in growth-permissive medium (Fig. 4), a structure generated by neural progenitor cells in vitro (4). As monolayers required significantly less manipulation prior to animal transplantation studies, all subsequent cells were prepared as monolayers.

Both immortalized (GuRt09) and nonimmortalized (GS076) human fetal retinal cells form neurospheres. Phase contrast images showing GS076 cells (A) and GuRt09 (C) cells in neurosphere formation in culture. Nonimmortalized (B) and immortalized (D, E) cells within the neurospheres expressed both nestin and Ki67. Antigens of interest are color coded; DAPI-stained nuclei are blue. Scale bars: (A, C) 200 μm; (B, D–E) 20 μm.
Transplanted Human Retinal Progenitor Cells Exhibit Limited Integration and Survival in the Rat Retina
The integration and survival of the GS076 and GuRt09 cell lines in the immunosuppressed rat retina were evaluated by immunohistochemical analysis using specific antibodies directed against human nuclear (HNA) and mitochondrial (HMIT) antigens. During the transplantation procedure, it was noted whether the cells were injected into the subretinal space or the vitreal cavity for each graft undertaken. Subpopulations of host microglia/macrophages may autofluoresce (Fig. 5A–D), possibly due to phagocytosis of autofluorescent debris zone material. This can lead to false-positive fluorescent signals when identifying grafted cells. We therefore developed a technique to designate incontrovertibly transplanted cells by identifying microglia/macrophages with an antibody directed against CD68 (ED1) and visualizing with a peroxidase reaction for bright-field analysis (Fig. 5). This approach did not interfere with the immunofluorescent detection of the human antigens.

Autofluorescence artifact in retinal transplants is produced by CD68-positive macrophage/microglia. This histological section (A–D) is a single confocal optical slice (<1 μm) taken through the injection site of an RCS dystrophic rat, 26 days postgrafting of nonimmortalized human fetal retinal cells. The macrophage/microglia are stained black with the ED1 antibody (anti-CD68) using a peroxidase reaction. When viewed under the confocal microscope this technique reveals populations of autofluorescent CD86-positive cells (large arrows) that may have phagocytosed autofluorescent debris zone material. Arrowheads point to CD68-positive, nonautofluorescent cells. Nonimmortalized human retinal progenitor cells transplanted into neonatal rat hosts were found attached to the host lens (arrow in E, shown at 2.5x magnification to the right). This material was taken 5 days postgrafting into the vitreous cavity of a postnatal day 3 host. The human cells were Ki67 negative (E) but expressed nestin (F). Inset of F shows DAPI staining 2x magnification. Grafted cells were in close proximity to CD68-positive cells. (A) and (B) show peroxidase staining on the Nomarski channel. Isolated confocal channels are shown to the right of images (E) and (F). Antigens of interest are color coded; DAPI-stained nuclei are blue. Outer nuclear layer (ONL); inner nuclear layer (INL); debris zone (dz). Scale bars: (A–D, F) 50 μm; (E) 200 μm.
At every time point examined there was an intimate association between CD68-positive host macrophage/microglia and the grafted human cells (Figs. 5–8). Following grafting of cells into neonatal (P3) hosts, human cells were only found in small clumps in the vitreous or on the surface of the lens (Fig. 5). In the adult dystrophic RCS rat only a small proportion of the 5 × 104 nonimmortalized human retinal cells transplanted into the subretinal space survived and by 5 days the remaining cells were closely associated with infiltrated CD68-positive host cells (Fig. 6A). In contrast to our findings in vitro, the surviving nonimmortalized cells, transplanted into the vitreous, did not express Ki67 at any of the time points studied (5, 26, and 63 days postgrafting), indicating that they remained largely in the resting phase of the cell cycle (Fig. 6C). Moreover, unlike the expression profile in vitro, the grafted cells were also negative for NF-160/NF-200 (markers of retinal ganglion cells) and recoverin/rhodopsin, a characteristic of photoreceptor (PR) cells (Fig. 6, Table 2). Where grafted nonimmortalized cells had inadvertently entered the vitreous, however, the cells survived in greater number and remained positive for nestin expression up to 26 days postgrafting.

Following transplantation into RCS dystrophic rat eyes, nonimmortalized (GS076) cells can survive and express nestin at the retinal–vitreous interface. Nonimmortalized cells grafted into the subretinal space showed poor staining for human markers 5 days postgrafting into the subretinal space (A). Strong positive staining for CD68 at 5 days postgrafting, with CD68-positive cells infiltrating/encapsulating the human graft (arrows in A, B). Surviving human cells were found on the vitreal aspect of host retina at 26 days postgrafting (B and arrows in C), where they were in close association with CD68-positive microglia/macrophages (C, D). The human cells were negative for Ki67 (C) but, as indicated by arrows in (D), they were positive for nestin (dashed box in D shown at higher magnification to the right). Grafted cells, in the vitreous, were negative for recoverin (E) and the ganglion cell marker neurofilament 200 (F). Isolated confocal channels are shown below or to the right of (C) and (D), respectively. (A–D) show peroxidase staining on the Nomarski channel. Antigens of interest are color coded; DAPI-stained nuclei are blue. Outer nuclear layer (ONL); inner nuclear layer (INL); ganglion cell layer (GCL). Scale bars: (A) 200 μm; (B, C) 100 μm; (D) 200 μm; (E) 50 μm.
Unlike the nonimmortalized cells, the GuRt09 cell line could be clearly identified in the subretinal space with human markers 5 days postgrafting (Fig. 7). The human cells were positive for Ki67 and, as with the GS076 cells, were surrounded by host CD68-positive phagocytes. The immortalized cells occasionally formed discrete clusters in the degenerating subretinal space where they expressed some nestin immunoreactivity while remaining largely negative for recoverin (Fig. 7A, B). Detailed analysis revealed that the immortalized human cells (but not host CD68-positive cells) were positive for rhodopsin immunoreactivity (Fig. 7D) but negative for cone-specific S-opsin (Fig. 7E).

At 5 days postgrafting into the eye of RCS dystrophic rats, immortalized (GuRt09) cells were present in the subretinal space. The grafted human cells only occasionally expressed nestin and were associated with CD68-positive cells (arrows in A). Immortalized grafted cells showed no obvious recoverin expression 5 days postgrafting into the dystrophic subretinal space (arrow in B). As shown in C (contents of dashed box shown at 2x magnification to the right), both grafted human cells and the CD68-positive microglia/macrophages were Ki67 positive. The grafted immortalized human cells were often found to form sphere-like structures in the subretinal space (D). The arrow in (D) shows one of these spheres associated with rhodopsin immunoreactivity in the debris zone (dz). This sphere is shown at 2.5x magnification to the right, where the separated channels of a single confocal optical slice (<1 μm) reveal rhodopsin-like immunoreactivity within the human cells. The cells formed spherical structures that were negative for the cone marker S-opsin (E). The dashed box in (E) is shown at 5x magnification to the right. Concurrent peroxidase staining revealed that the grafted human cells were infiltrated and surrounded by CD68-positive microglia/macrophages. Isolated confocal channels are shown to the right of each image. Antigens of interest are color coded; DAPI-stained nuclei are blue. Outer nuclear layer (ONL); inner nuclear layer (INL); debris zone (dz). Scale bars: (A–D) 50 μm; (E) 100 μm.
At 9 weeks postgrafting there was no evidence of GS076 cell survival in either the subretinal space or the retinal parenchyma. However, at this late time point immortalized GuRt09 cells survived at the interface between the retina and the vitreous (small arrows in Fig. 8A) but were not found in the subretinal space. At 13 weeks of age the outer nuclear layer (containing the cell bodies of rods and cones) of the dystrophic retina is reduced to a layer of 1–2 cells thickness (Fig. 8B). However, in GuRt09-grafted animals the outer nuclear layer was preserved (large arrows in Fig. 8A) despite the absence of human cells in the subretinal space. The surviving immortalized cells were still associated with some host CD68-positive cells and were occasionally positive for nestin but remained negative for other markers including NF-160 and NF-200 (Fig. 8E, F, G, Table 2). It is important to note that although a proportion of grafted cells were positive for the proliferation antigen Ki67, we did not see an increase in the absolute number of human cells or tumor-like formations occurring. The results of cell survival for the different transplant groups are summarized in Table 3. In addition to the transplantations of single cell suspensions, we also grafted the nonimmortalized and immortalized cells in the form of neurospheres (n = 4 for each group). At 9 weeks postgrafting there were no signs of cell survival (data not shown), indicating that grafting neurospheres does not enhance cell survival or integration.

Immortalized human fetal retinal cells (GuRt09) survive for 9 weeks postgrafting into the RCS dystrophic eye. Nissl-stained retinal sections revealed a preservation of the outer nuclear layer (large arrows in A) that was associated with large cells lining the vitreal surface of the retina (small arrows in A). (B) A retinal section from a 13-week-old dystrophic rat; note the reduction of the outer nuclear layer to almost a single layer of cells (arrows in B). The immortalized human cells were associated with CD68-positive microglia/macrophages (2x magnification of dashed box in C) and were positive for Ki67 (D). Grafted human cells were negative for neurofilament 160 (E), neurofilament 200 (F), and only occasionally expressed nestin (G). Isolated confocal channels are shown to the right of each image. Antigens of interest are color coded; DAPI-stained nuclei are blue. (B) and (E) show peroxidase staining on the Nomarski channel. Inner plexiform layer (IPL); inner nuclear layer (INL); ganglion cell layer (GCL). Scale bars: (A) 200 μm; (B) 100 μm; (C-G) 50 μm.
A Summary of Transplantation Data

Summary of transplant data with survival times postgrafting and experimental outcomes. Experiments are divided into short- and long-term survival groups. If grafts were present then this is indicated by “yes” and the number of animals in which this was the case is reported within the parentheses. Grafts were either found in the vitreous or subretinal space (SRS). In each condition of the short-term group, n = 4 animals were sacrificed at 5 days and those containing a graft in the subretinal space are highlighted by an asterisk.
Discussion
Human retinal progenitor cells at 12 weeks of development are partially committed towards a retinal phenotype and hence competent to develop into both cone and rod photoreceptors, the latter being one of the last neuronal cell types to develop in the mammalian retina (23). The early phase of rod photoreceptor (PR) differentiation begins at the foveal edge at 10.5 weeks and by weeks 11–12 these rod cells begin to express specific rod PR markers, including recoverin (9). Previous studies have shown that retinal cells extracted at the same time point may differentiate into several retinal cell phenotypes (36), indicating at least some multipotentiality. By harvesting and immortalizing cells at this developmental stage we have created a renewable and stable cell line that may retain the potential to differentiate into PR cells, a key goal in cell transplantation strategies for treating retinal degenerative diseases. Under in vitro cell culture conditions, both the nonimmortalized and immortalized cell lines expressed similar proportions of progenitor and maturation markers (such as NF-160 and NF-200) throughout the 10 or 32 passages over which they were cultured, respectively. This demonstrates that the immortalized cell line has the capacity to self-replicate and retain progenitor status over multiple cell doublings. Nevertheless, although clonally derived, the GuRt09 cells also displayed various characteristics with a proportion of cells exhibiting further differentiation markers.
Until recently the view has been that immature PR progenitor cells may be best placed to integrate and differentiate in the adult retina. Recent studies, however, have challenged this view as integration into the correct anatomical location in the adult retina appears to be best achieved with PR precursor cells (21,33). In future studies, therefore, it may be necessary to introduce extrinsic cues or genes (such as Nrl or Crx) to drive the immortalized retinal progenitor cells towards their final phenotype (rods or cones) and to harvest these cells prior to grafting.
Having established that we could generate an immortalized cell line with an in vitro profile comparable to nonimmortalized cells, we next determined its capacity to integrate and differentiate in the rat retina. First we established an histological technique that provided us with a definitive measure of grafted cells whereby we costained for host microglia/macrophages that can autofluoresce giving potentially false-positive signals and human nuclear/mitochondrial markers. In past studies this problem has been neglected, leading to the possible false identification of transplanted cells. In the developing neonatal rat (P3) both immortalized and nonimmortalized cells grafted into the vitreous failed to integrate into the neural retina and were only observed in the vitreous or on the surface of the lens. This may indicate the different immunological mechanisms at play in the subretinal space versus the vitreous cavity. The likely failure to integrate may in part be due to the location of the graft and the presence of the inner limiting membrane (ILM) preventing cell migration. The ILM comprises a basement membrane and the foot processes of Müller glia cells from which it is produced, and has previously been reported to hinder the passage of cells from the vitreous to the neural retina (39). Furthermore, the basement membrane provides a suitable surface for preferential attachment thus preventing further migration.
It has previously been reported that integration of stem/progenitor cells into the normal adult retina is limited but can be enhanced if the retina is lesioned (5,37), suggesting that factors released during retinal damage promote stem cell migration and possibly differentiation. However, despite considerable evidence that the damaged or diseased CNS enhances stem cell integration (27), there is convincing contrary evidence that in the retina reactive gliosis occurring as a consequence of disease or damage can inhibit this process (13). When grafted into the subretinal space of the RCS rat, our cell lines showed little evidence of any integration into the PR layer. As with the ILM, the outer limiting membrane (OLM) also represents a physical barrier to integration, and recently strategies to disrupt this anatomical barrier have been proposed to facilitate integration (33). Nevertheless, even in the adult WT retina and in the presence of an intact OLM limited integration of allogeneic PR precursor cells has been observed (21), suggesting that failure of the human progenitor cells to show any evidence of integration is likely to be a consequence of either immune-mediated rejection of the xenograft or the immature state of differentiation. As no integration was recorded in congenic RCS rats, failure to integrate in the dystrophic animals is unlikely to be due simply to glial scarring. It will be of interest, therefore, to determine whether further in vitro differentiation prior to grafting enhances their capacity to integrate. Despite poor integration, immortalized cells survived in the subretinal space for a limited period and exhibited more cell division than their nonimmortalized counterparts. The active proliferation of the immortalized cells was not, however, associated with tumor formation up to 9 weeks postgraft, suggesting that the cell division observed was a consequence of the cells remaining in an undifferentiated progenitor state. This was supported by the observation that immortalized grafted cells occasionally expressed nestin.
The lack of further differentiation of the grafted cells suggests that the environmental cues needed to drive differentiation are missing in the adult retina. Additionally, inhibitory signals may also be present, enforcing the quiescent milieu. Such inhibitory signals may be intrinsic to the mature retina but may also be induced through the pathology. For example, the close association between host macrophage/microglial cells and donor cells may be partly responsible for the lack of differentiation of the latter, as activated microglia secrete IL-6, which limits progenitor cell turnover and differentiation towards postmitotic retinal cells (8).
The enhanced survival of photoreceptors in dystrophic RCS rats over the site of the graft mirrors observations made in other cell transplantation studies (6,17,18,20,30,34). At the 9-week postgrafting survival time, the extent of the surviving outer nuclear layer was matched by the extent of the surviving immortalized cell graft, in particular at the border between the retina and vitreous. The ability of the grafted cells to rescue degenerating retinal photoreceptors is consistent with findings in previous studies. It has been proposed that the enhanced survival of PR cells may occur through a paracrine mechanism of the grafted cells (17,18,20,34), highlighting the potent neuroprotective effects of the grafted cells. Additionally, the stimulation of macrophages to the site may also assist in clearing the debris zone, thus improving nutrient availability and waste product clearance.
In summary this proof-of-concept study has demonstrated that immortalized human retinal progenitor cells are capable of self-replication in vitro and are able to retain their progenitor status. Cell transplantation studies did not demonstrate either differentiation or integration into host retina with either of the cell lines tested. However, despite limited survival of the grafted human cells, an observation of cells exhibiting a neuroprotective effect was observed in only one of the grafts in the degenerating retina of the RCS rat. Furthermore, immortalized retinal progenitor cells were not tumorigenic over the period studied. Further studies are required to elucidate if enhanced integration and photoreceptor differentiation is a viable goal in cell therapy for patients with degenerative retinal disease. If patients were treated at the early onset of disease, it is envisaged that a neuroprotective strategy could significantly slow the progression. Immortalized retinal progenitor cells may therefore provide a renewable, stable, and consistent supply of transplantable cells that, following thorough characterization and compliance with regulatory authorities, may be evaluated for the treatment of retinal degeneration.
Footnotes
Acknowledgment
This work was supported by grants from the Medical Research Council, UK.