
Abstract
Multipotent adult progenitor cells (MAPCs) are bone marrow-derived nonhematopoietic stem cells with a broad differentiation potential and extensive expansion capacity. A comparative study between human mesenchymal stem cells (hMSCs) and human MAPCs (hMAPCs) has shown that hMAPCs have clearly distinct phenotypical and functional characteristics from hMSCs. In particular, hMAPCs express lower levels of MHC class I than hMSCs and cannot only differentiate into typical mesenchymal cell types but can also differentiate in vitro and in vivo into functional endothelial cells. The use of hMSCs as cellular immunomodulatory stem cell products gained much interest since their immunomodulatory capacities in vitro became evident over the last decade. Currently, the clinical grade stem cell product of hMAPCs is already used in clinical trials to prevent graft-versus-host disease (GVHD), as well as for the treatment of acute myocardial infarct, ischemic stroke, and Crohn's disease. Therefore, we studied the immune phenotype, immunogenicity, and immunosuppressive effect of hMAPCs in vitro. We demonstrated that hMAPCs are nonimmunogenic for T-cell proliferation and cytokine production. In addition, hMAPCs exert strong immunosuppressive effects on T-cell alloreactivity and on T-cell proliferation induced by mitogens and recall antigens. This immunomodulatory effect was not MHC restricted, which makes off-the-shelf use promising. The immunosuppressive effect of hMAPCs is partially mediated via soluble factors and dependent on indoleamine 2,3-dioxygenase (IDO) activity. At last, we isolated hMAPCs, the clinical grade stem cell product of hMAPCs, named MultiStem, and hMSCs from one single donor and observed that both the immunogenicity and the immunosuppressive capacities of all three stem cell products are comparable in vitro. In conclusion, hMAPCs have potent immunomodulatory properties in vitro and can serve as a valuable cell source for the clinical use of immunomodulatory cellular stem cell product.
Keywords
Introduction
Multipotent adult progenitor cells (MAPCs) are postnatal nonhematopoietic adherent stem cells, which originally have been isolated from the bone marrow copurified with mesenchymal stem cells (MSCs) (7). A recent comparative analysis between human MAPCs (hMAPCs) and human MSCs (hMSCs) has shown that hMAPCs and hMSCs are two distinct cell populations. These cells differ in terms of phenotype, expansion, and differentiation potential (25). Human MAPCs could be expanded for a significantly longer time than hMSCs. Roobrouck et al. reported that hMAPCs can be expanded for more than 30 passages (>70 PDs) in contrast to hMSCs, which can only be maintained for 12–18 passages (~20–25 PDs). The extended expansion capacity was consistent with a higher and more sustained telomerase activity in hMAPCs. Human MAPCs are negative for hematopoietic [cluster of differentiation 34 (CD34), CD45, and v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (CD117; c-kit)] and endothelial [kinase insert domain receptor (CD309; KDR), CD34] cell surface markers. They do also not express CD56 and CD271 and are dimly positive for CD146. On the other hand, hMAPCs express high levels of CD44, CD13, CD73, CD90, and CD105. In addition, in contrast to hMSCs, hMAPCs are negative for CD140a, CD140b, and alkaline phosphatase (ALP) and express major histocompatibility complex (MHC) class I at lower levels. hMAPCs can differentiate like hMSCs into typical mesenchymal cell types, including adipocytes, osteoblasts, and chondrocytes in vitro, while only hMAPCs can differentiate into functional endothelium in vitro and in vivo. Whether hMAPCs and hMSCs are derived from the same cell type in vivo is not yet known. However, Roobrouck et al. showed that the differences in phenotype, proliferation capacity, differentiation ability, and even gene expression profile between hMAPCs and hMSCs were partially due to differences in culture conditions and were interchangeable (25). We have as such no proof that the hMAPC culture conditions preserve more efficiently the undifferentiated state of these cells in vitro compared to the conditions used for MSCs.
There are also differences between human and rodent MAPCs. It has been published that rodent MAPCs express high levels of the pluripotency transcription factor octamer binding transcription factor 4 [Oct4] (30), whereas hMAPCs are known to express low levels of OCT3A (25).
Similar to hMSCs, the clinical use of hMAPCs is being explored in different disease conditions. hMSCs were first studied in the context of tissue repair (10, 27), but subsequently the use of hMSCs gained interest in the field of hematopoietic stem cell transplantation to prevent rejection and enhance hematopoietic engraftment (15). Coinfusion of hMSCs at the time of allogeneic hematopoietic stem cell transplantation has been shown to be feasible and safe (8, 11, 16). However, the most important clinical application today is based on their strong immunosuppressive capacities in vitro (21, 29). In particular, hMSCs suppress T-cell activation and proliferation induced by alloantigens and mitogens in vitro. This suppression is without any MHC restriction as it can be mediated by both autologous and allogeneic MSCs (1, 4, 20). However, the mechanism of suppression of hMSCs on allogeneic T-cell proliferation is not yet completely understood (31). It is generally accepted that it is mediated via both contact dependent and soluble factors. Nowadays, based on this strong in vitro immunomodulatory capacities, hMSCs are applied to modulate graft-versus-host disease (GVHD) in vivo (12, 14), and their use is currently being explored in different autoimmune diseases, like Crohn's disease and rheumatoid arthritis.
One of the major drawbacks of hMSCs is their limited proliferative capacity such that large clinical trials cannot be done using well-characterized clinical grade single-donor cell products. This becomes particularly important when off-the-shelf use of stem cells is considered for treatment of acute medical conditions such as stroke, myocardial infarction, and peripheral ischemia. As indicated above, the more extensive proliferation and differentiation ability of hMAPCs may circumvent these problems. Therefore, procedures were developed for large-scale expansion of hMAPCs under Good Manufacturing Practice (GMP) conditions, and this clinical grade stem cell product of hMAPCs is called MultiStem (2). This allows for the creation of a master cell bank, from which working cell banks can be generated to treat patients with a uniform single-donor cell product that has undergone extensive safety testing and characterization. However, as these cellular products will be used off-the-shelf for treatment in allogeneic patients, their immunological behavior should be evaluated.
hMAPCs are now emerging as advanced therapy medicinal products for clinical practice, and so we have studied the immunogenicity of hMAPCs in the context of their off-the-shelf use in acute diseases aiming at tissue regeneration, as well as their immunosuppressive behavior and the mechanism of suppression in the context of deleterious immune responses and inflammation. In addition, we compared at the end the immunogenicity and immunomodulatory capacity of hMAPCs with hMSCs derived from the same donor. The data of this study are important to document safety and immunological potency of hMAPCs before their widespread use in clinical practice is initiated.
Materials and Methods
Isolation and Culture of Stem Cells
Human MAPC (n = 2) isolations were performed by the Stem Cell Institute Leuven (SCIL) from bone fragments of children (10 and 12 years old) undergoing orthopedic surgery after obtaining informed consent in accordance with the guidelines of the Medical Ethics Committee of the University Hospitals Leuven. Single-donor triplets of hMAPCs, hMSCs, as well as MultiStem were isolated by Athersys/ReGeneys from bone marrow of a 30-year-old female and twice from a 45-year-old male volunteer, again with informed consent and ethical approval.
Isolation and culture of the cells was carried out as previously described (25, 30). Briefly, hMAPCs were generated by plating the total cell fraction at 0.5×106 cells/ cm2 in a medium consisting of 60% Dulbecco's modified Eagle's medium (DMEM) low glucose (Gibco, Invitrogen, Carlsbad, CA, USA), 40% MCDB-201 (Molecular, Cellular and Developmental Biology, University of Colorado, Boulder, CO, USA; media-201, Sigma-Aldrich, St. Louis, MO, USA), supplemented with 50 nM dexamethasone, 10−4 M l-ascorbic acid, 1×selenium–insulin–transferrin (ITS), 0.5×linoleic acid–bovine serum albumin (LA-BSA) (all from Sigma-Aldrich), 1% penicillin/streptomycin (Gibco, Invitrogen), along with 2% serum supreme (Lonza BioWhittaker, Basel, Switzerland), and 10 ng/ml human platelet-derived growth factor-BB (PDGF-BB; R&D Systems, Minneapolis, MN, USA) and epidermal growth factor (EGF; Sigma-Aldrich). MAPC cultures were maintained under hypoxic conditions (5% O2) at a density of 400 cells/cm2 and were split every 2–3 days. Clonal populations of hMAPCs isolated by SCIL were obtained through limiting dilution by plating five cells per well in a 96-well or 48-well plate (Corning, NY, USA) between passages 5 and 10. MultiStem, the large-scale expanded clinical grade stem cell product, was isolated from the bone marrow using similar isolation protocol and media as hMAPCs, except that MultiStem was cultured with 18% fetal bovine serum (FBS; Lonza, Verviers, Belgium), plated at a density of 2,000 cells/cm2, and was not clonally derived. Cells were used at passages 5–14.
Human MSCs were generated from bone marrow by plating the mononuclear fraction, obtained after Ficoll density gradient centrifugation (Lymphoprep, Axis-Shield ProC As, Oslo, Norway), at 0.5×106 cells/cm2 in MSC growth medium (Lonza) containing DMEM high glucose, 10% FCS (Lonza), and 100 IU/ml penicillin (Lonza) and 100 μg/ml streptomycin (Lonza). MSC cultures were maintained at 5,000 cells/cm2, at normal oxygen level (20% O2), were split every 4–7 days, and were not clonally derived. Cells were used at passages 8–9.
Phenotypic analysis of the stem cells was done using fluorochrome-conjugated antibodies recognizing CD34, CD45, c-kit, CD56, CD271, CD44, CD13, CD73, CD90, CD146, CD140b, CD140a (BD Pharmingen, Erembodegem, Belgium), and CD105 (R&D Systems). Human MAPCs, hMSCs, and MultiStem were negative for CD34, CD45, c-kit, CD56, and CD271 and were all positive for CD44, CD13, CD73, CD90, CD105, and CD146. In contrast to hMAPCs and MultiStem, hMSCs are positive for CD140b and express higher levels of human leukocyte antigen (HLA)-ABC (BD Pharmingen) and CD140a compared with hMAPCs and MultiStem (25).
Peripheral Blood Mononuclear Cells and T-Cells
All subjects donating blood for these experiments were healthy volunteers of both sexes, aged 20–50 years. Peripheral blood mononuclear cells (PBMCs) were separated by Ficoll-Hypaque density gradient centrifugation (specific density, 1.077 g/ml). T-cells were further purified using two rounds of a complement-mediated depletion of all non-T-cells with lympho-KWIK-T (One λ, Los Angeles, CA, USA). Briefly, the T-lympho-KWIK reagent (0.8 ml) was added to 20–30×106 PBMCs, and the mixture was incubated for 1 h at 37°C. Cells were centrifuged at 1,845×g for 2 min and washed twice. A mixture of complement-fixing antinatural killer (NK) cell monoclonal antibody (mAb) of the IgM subclass (BD Biosciences, San Jose, CA, USA) was added to the cells. The cells were incubated for 30 min at 4°C and washed once. Incubation of T-lympho-KWIK reagent was then repeated. Cells were again centrifuged at 1,845×g for 2 min, washed twice, and resuspended in culture medium Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% autologous plasma, 2 mmol/l L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Lonza). The purified T-cell population contained at least 97% CD3+ cells (BD Pharmingen) as determined by flow cytometry.
T-Cell Response Upon Allogeneic hMAPCs
Responder purified T-cells (1×105) were stimulated with 5×104 allogeneic, irradiated (30 Gy) hMAPCs or CD2-depleted PBMCs (see below) as control in one well of a flat-bottomed 96-well plate (Greiner Bio-One, Wemmel, Belgium). For each experiment, all stimulator cells were isolated from the same donor. In some experiments, hMAPCs were incubated with 100 U/ml interferon (IFN)-γ (Roche, Vilvoorde, Belgium) for up to 48 h in their respective culture medium and oxygen concentration prior to coculture with responder T-cells in order to upregulate MHC class I and MHC class II. T-cell proliferation was measured at day 6 by means of 8-h incubation with 1 μCi/well [3H]thymidine (ICN Biomedicals, Costa Mesa, CA, USA). [3H]Thymidine incorporation was measured by using a liquid scintillation counter [Tri-Carb® 2100TR Liquid Scintillation Counter (PerkinElmer Life Sciences, Inc., Zaventem, Belgium)]. The data were analyzed as mean counts per minute (cpm) of triplicate or quadruplicate wells. The results are expressed as stimulation index. Stimulation index (SI) was calculated as follows: [cpm (stimulated cells) – cpm (unstimulated responders cells)] / cpm (unstimulated responder cells). In order to evaluate the presence of activation markers on responder CD3+ cells by flow cytometry, stimulator PBMCs were depleted of CD2+ cells using Dynabeads® CD2 Pan T (Invitrogen, Merelbeke, Belgium), according to manufacturer's protocol.
Immunosuppression by hMAPCs
For the mixed lymphocyte reactions (MLR), 1×105/well responder T-cells or unfractionated PBMCs were mixed with 1×105/well irradiated (30 Gy) allogeneic PBMCs in 200 μl of culture medium in a flat-bottomed 96-well plate (Greiner Bio-One). T-cells were incubated for 6 days at 37°C and 5% CO2. Irradiated (30 Gy) autologous, allogeneic, or third-party hMAPCs were added at different suppressor/responder (S/R) ratios. T-cell proliferation was measured at day 6 by means of an 8-h pulse with 1 μCi/ well [3H]thymidine. [3H]Thymidine incorporation was measured by using a liquid scintillation counter. The data were analyzed as mean counts per minute (cpm) of triplicate or quadruplicate wells. The results are expressed as percentage of response related to the control response in the absence of stem cells.
In some experiments, 4.5 Lf/ml tetanus toxoid (NIBSC, Hertfordshire, Belgium) or 1 μg/ml phytohemagglutinin (PHA, Murex, Dartford, England) were used to induce T-cell proliferation in the presence or absence of hMAPCs. For this, unfractionated PBMCs were used as responder cells.
To analyze the involvement of indoleamine 2,3-dioxygenase (IDO), prostaglandin E2 (PGE2), transforming growth factor (TGF)-β, and interleukin (IL)-10 as immunosuppressive mediators, we used their respective specific inhibitors: 200 μM 1-methyl-tryptophan (1-MT; Sigma-Aldrich), 2 μg/ml indomethacin (Cayman Chemicals Company, Ann Arbor, MI, USA), 50 μg/ml anti-TGF-β neutralizing mAb (R&D Systems, Abingdon, UK), and 2.5 μg/ml anti-IL-10 plus 2.5 μg anti-IL-10 receptor mAb (both from R&D Systems).
In some experiments, T-cells were stimulated with allogeneic irradiated PBMCs in the presence or absence of hMAPCs (S/E 1/2). After a 2-day resting period, these T-cells were restimulated in a secondary MLR for 4 days with freshly isolated irradiated PBMCs in the absence or presence of hMAPCs.
Transwell Cultures
Transwell chambers with a 0.2-μM Anapore® membrane (Nunc, Rochester, NY, USA) were used to separate hMAPCs from the MLR via a semipermeable membrane. The MLR was performed in the lower well of a 96-well plate as described. Human MAPCs were placed in the inner transwell chamber. After 6 days, the inserts were removed and the T-cells were pulsed with 1 μCi/well [3H] thymidine for 8 h.
Flow Cytometry
Responder cells (1 × 106) and irradiated (30 Gy) allogeneic stimulator cells (1 × 106; single-donor CD2-depleted PBMCs or hMAPCs) were mixed in a final volume of 1 ml in a 24-well plate (Greiner Bio-one). After 6 days, responder cells were harvested and surface staining was performed according to the manufacturer's instructions and modified as follows: 1-2 × 105 T-cells per sample were suspended in 100 μl staining buffer. Nonspecific staining was blocked with human autologous serum and cells were subsequently surface stained with 5 μl fluorescence-conjugated specific mAb anti-CD3, anti-CD4, anti-HLA-DR, anti-CD25, and anti-inducible T-cell costimulator (CD278; ICOS) (BD Pharmingen).
For the analysis of the immune phenotype fluorescence-conjugated specific mAb anti-HLA ABC, anti-HLA DR, anti-CD80, anti-CD86, anti-CD40, anti-CD54 (eBioscience, Inc., San Diego, CA, USA), and anti-CD58 (BD Pharmingen) were used. The immune phenotype was studied before and after 48 h of stimulation with 100 U/ml IFN-γ (Roche). Isotype control staining was performed. Cells were gently mixed and incubated for 30 min at 4°C. Intracellular staining for forkhead box P3 (FoxP3) was performed according to the manufacturer's protocol. Alexa Fluor® 488-conjugated anti-FoxP3 was purchased from eBioscience (eBioscience, Inc.).
Acquisition was done by using a FACSort or a FACSCanto (BD Biosciences). For analysis of the samples, CellQuest (BD Biosciences) or FlowJo (Tree Star, Inc., Ashland, OR, USA) software was used.
Cytokine Production and Cytometric Bead Array Assay
Supernatants were taken at day 6 of the coculture of T-cells with allogeneic hMAPCs. Cytokines (IFN-γ, IL-13, and IL-10) were determined by cytometric bead array (CBA) Flexset (BD Biosciences) and IL-5 via ELISA (BD Biosciences) according to manufacturer's instructions. Production of cytokines by hMAPCs after 6 days of culture [IFN-γ, tumor necrosis factor (TNF)-α, IL-10, IL-2] was measured by CBA Flexset (BD Biosciences).
Determination of Kynurenine Production
IDO activity was detected by determining the IDO-catalyzed tryptophan to kynurenine degradation. Kynurenine in culture supernatants was measured photometrically as described (3). Human MAPCs and MultiStem were cultured in the presence of IFN-γ (R&D) ± TNF-α (Sanquin, Amsterdam, The Netherlands) ± IL-1β (Gentaur, Brussels, Belgium) (each 100 U/ml) for 3 days in tryptophan-supplemented medium (100 μg/ml tryptophan, Sigma-Aldrich) before determination of kynurenine production. Experiments were performed in duplicate in 10 different 96-well plates, which were pooled afterwards per condition for performing the read-out in triplicate.
Statistical Analysis
Statistics were calculated with Prism software 5.0 (GraphPad Software, Inc., San Diego, CA, USA). For comparisons between two groups, paired or unpaired t tests were used. For comparison to a known value, one-sample t test was used. Statistical significance was calculated by one-way ANOVA with Dunnett's post hoc test, Bonferroni's post hoc test, or two-way ANOVA for multiple comparisons.
Results
Human MAPCs Are Nonimmunogenic for Allogeneic T-Cells
Human MAPCs expressed low levels of MHC class I (HLA ABC), and were negative for MHC class II (HLA DR), the costimulatory molecules CD80, CD86, and CD40, and the adhesion molecule intracellular adhesion molecule (ICAM-1; CD54) and CD58. Upon stimulation with IFN-γ, expression of MHC class I, class II and ICAM-1 was induced, whereas the expression of CD80, CD86, and CD40 remained unchanged (Table 1). The upregulation of MHC class I and ICAM-1 was already present at 24 h, although not significant due to the variable expression, whereas the upregulation of MHC class II was only induced upon 48-h stimulation. Incubation of hMAPCs with IFN-γ during 72 h did not further enhance expression of MHC class I, MHC class II, and ICAM-1 (data not shown).
Immune Phenotype of hMAPCs

Flow cytometric analysis of human multipotent adult progenitor cells (hMAPCs) for antigen-presenting molecules [major histocompatibility complex (MHC) class I, MHC class II], adhesion molecules [cluster of differentiation 54 (CD54), CD58], and costimulatory molecules (CD80, CD86, and CD40) before and after 48-h treatment with interferon (IFN)-γ. Results are expressed as mean ± SEM median fluorescence intensity of six independent experiments. Statistical significance was calculated with paired t test. FITC, fluorescein isothiocyanatae; PE, phycoerythrin.
To assess whether hMAPCs could induce a T-cell response in vitro, purified responder T-cells were stimulated with irradiated, allogeneic single-donor hMAPCs and CD2-depleted PBMCs as a positive control. Stimulator cells were cocultured with T-cells at a responder/stimulator ratio of 2:1. After 6 days, T-cell proliferation was measured by means of [3H]thymidine incorporation. T-cells responded robustly to the allogeneic CD2-depleted PBMC fraction. In contrast, hMAPCs induced a negligible proliferative response when cocultured with allogeneic T-cells (Fig. 1A). In addition, the expression of activation markers on responder CD3+ T-cells was evaluated upon coculture with hMAPCs. Five to 10% CD3+ cells expressed the activation markers CD25, HLA-DR, and inducible costimulator (ICOS) in response to allogeneic hMAPCs, whereas the response to allogeneic CD2-depleted PBMC was obvious (50–70%) (Fig. 1B). As compared to unstimulated T-cells, there is a slight increase of activation markers on responder T-cells upon allogeneic stimulation with hMAPCs. Although the induction of activation markers is lower on T-cells after stimulation with hMAPCs as compared to T-cells stimulated with CD2-depleted PBMCs, this still may reflect a low level of T-cell activation upon stimulation with allogeneic hMAPCs.

T-cell responses to allogeneic hMAPCs. (A) T-cell proliferation upon stimulation with allogeneic irradiated (30 Gy) single-donor cluster of differentiation 2 (CD2)-depleted peripheral blood mononuclear cells (PBMCs) and human multipotent adult progenitor cells (hMAPCs). Results are expressed as mean stimulation index ± SEM (n = 5). Statistical significance (**p < 0.01) compared to control with CD2-depleted PBMCs was tested with unpaired t test. (B) Flow cytometric analysis of activation markers on unstimulated T-cells or on T-cells stimulated for 6 days with allogeneic single-donor CD2-depleted PBMCs or hMAPCs. Results are representative of three independent experiments. (C–F) T-cells were cultured with irradiated allogeneic single-donor CD2-depleted PBMCs or hMAPCs. Supernatants were collected at day 6 and assessed for the indicated cytokines by cytometric bead array (CBA) or ELISA. Data are expressed as mean ± SEM pg/ml from three independent experiments. CD4, cluster of differentiation 4; HLA-DR, human leukocyte antigen-DR; ICOS, inducible T-cell costimulator; IFN-γ, interferon-γ; IL-5, interleukin-5.
To investigate whether the lack of T-cell response was due to the induction of suppressive regulatory T-cells (Tregs), the presence of CD4+CD25highFoxp3+ cells was determined after coculture of T-cells with hMAPCs. Flow cytometric analysis did not show an induction of Tregs by hMAPCs (4.6% CD4+CD25highFoxp3+ cells vs. 33% in control MLR and 3.9% in unstimulated T-cells) (data not shown). Further analysis of the supernatant from the coculture of T-cells with hMAPCs showed no production of Th1 (Fig. 1C) or Th2 (Fig. 1D, E) cytokines. In addition, the absence of proliferation upon stimulation of T-cells with hMAPCs is not related to the overproduction of IL-10, a known immunosuppressive cytokine (Fig. 1F).
MAPCs Suppress T-Cell Proliferative Responses
In the next series of experiments, the immunosuppressive properties of hMAPCs were investigated. Human MAPCs were added at different ratios to a primary MLR. There was a significant reduction in T-cell proliferation when responder T-cells (1×105/well) were stimulated with irradiated allogeneic PBMCs (1×105/well) in the presence of irradiated hMAPCs. This suppressive effect was dose dependent and most pronounced at the highest S/R ratios (Fig. 2A). Of note, in this series of 24 similar experiments, we could not detect a quantitative or qualitative difference between the MAPCs generated from children or adults.

Inhibitory effect of hMAPCs on allogeneic T-cell proliferation. (A) Purified T-cells were stimulated with allogeneic irradiated PBMCs in the presence and absence of different numbers of irradiated hMAPCs. The data are expressed as mean (±SEM) percentage proliferation as compared to a control mixed lymphocyte reaction (MLR) in the absence of hMAPCs, results are from 24 independent experiments. Statistical significance was tested when compared with control MLR. ANOVA with Dunnett's post hoc test, ***p < 0.001, **p < 0.01. (B, C) T-cells were stimulated with allogeneic irradiated PBMCs in the presence or absence of irradiated hMAPCs, either matched with responder cells (B) or used as third-party cells (C) at S/R ratios from 1:1 to 1:100. Data are expressed as mean percentage of proliferation ± SEM as compared to the MLR in the absence of hMAPCs from four independent experiments. ANOVA with Dunnett's post hoc test, ***p < 0.001, **p < 0.01.
Human MAPCs are intended to be used as an off-the-shelf product regardless of MHC compatibility with the patient. Therefore, we next questioned whether MHC compatibility of the stem cells with responding T-cells is mandatory. Human MAPCs were matched with responder cells or used as third-party cells. The suppressive activity of hMAPCs was independent of MHC matching (Fig. 2B, C). Besides, MHC matching of hMAPCs with stimulator cells equally suppressed the response of the responder T-cells (data not shown).
Immunosuppression by exogenous MAPCs will also aim to suppress an ongoing immune response; therefore, we next investigated whether delayed addition of hMAPCs to a primary MLR could also suppress T-cell proliferation. Therefore, irradiated hMAPCs were added at different S/R ratios at the initiation of or at day 3 of the MLR. We demonstrated that hMAPCs equally and dose-dependently suppressed an ongoing immune response (Fig. 3A).

Suppression of effector and memory T-cells by hMAPCs. (A) T-cells were stimulated with allogeneic irradiated PBMCs in the presence or absence of allogeneic hMAPCs either added at the initiation (day 0) of the MLR or at day 3 of a 6-day MLR. Data are expressed as mean percentage proliferation ± SEM of three independent experiments. Two-way ANOVA showed no significant main effect of time (p > 0.05). The main effect of S/R ratio was significant (p < 0.0001). The interaction was not significant (p > 0.05). (B) Human MAPCs were added during a primary MLR or a secondary MLR at S/R ratios of 1:1 and 1:10. The data are expressed as mean (±SEM) percentage proliferation as compared to control MLR in the absence of hMAPCs, and results are from three independent experiments. Two-way ANOVA showed no significant main effect of primary versus secondary MLR (p > 0.05). The main effect of S/R ratio was significant (p < 0.0001). The interaction was not significant (p > 0.05). (C, D) Unfractionated PBMCs were stimulated with the recall antigen tetanus toxin (TT; C) or phytohemagglutinin (PHA; D) in the absence or presence of hMAPCs at S/R ratio of 1:2. T-cell proliferation for TT- and PHA-stimulated cultures was performed respectively at day 6 and day 4. Results are expressed as mean percentage proliferation ± SEM of respectively two and five independent experiments. Statistical significance was calculated with the one-sample t test, *p < 0.05, **p < 0.01.
Furthermore, immunosuppression was investigated in the context of effector T-cell responses. For this, suppression assays were performed at the time of secondary MLR between T-cells and irradiated allogeneic PBMCs. Human MAPCs were still able to dose-dependently suppress the T-cell response during a secondary MLR (Fig. 3B). This suppression of hMAPCs was as effective as the suppression of a primary MLR. Finally, immunosuppression of a memory response was studied using the model of T-cell proliferation upon tetanus toxin antigen stimulation. The memory T-cell response on this recall antigen was also strongly suppressed by hMAPCs (Fig. 3C). As an additional condition, similar inhibitory effects on T-cell proliferation were observed when responder PBMCs (1×105/well) were stimulated with the polyclonal stimulus, PHA (Fig. 3D).
Pretreatment with IFN-γ Did Not Alter the Suppressive Effect of hMAPCs on T-Cell Proliferation
Off-the-shelf stem cells may be used in proinflammatory conditions, which theoretically might alter the phenotype and function of hMAPCs. We initiated a series of experiments in which hMAPCs were pretreated with IFN-γ for 48 h. After culturing in the presence of IFN-γ, the MHC class I molecules were upregulated and the expression of MHC class II molecules was detected on hMAPCs (Table 1). The adhesion molecule ICAM-1 was also upregulated. In spite of these phenotypic changes, the immunogenicity of IFN-γ pretreated hMAPCs was not enhanced (Fig. 4A). In addition, IFN-γ-pretreated hMAPCs were tested for their immunosuppressive effect on T-cell proliferation. Similarly, the immunosuppressive potency of IFN-γ-pretreated hMAPCs was not altered in the MLR model (Fig. 4B). However, IDO activity in two different isolations of hMAPCs was induced upon stimulation with IFN-γ, since kynurenine, the catabolite of tryptophan, in culture supernatants derived from hMAPCs incubated with IFN-γ was detected. The induction of IDO activity was quite specific for IFN-γ, because unstimulated cells or cells stimulated with TNF-α or IL-1β alone or in combination showed no or only very limited induction of IDO activity. There was an additional effect of TNF-α in combination with IFN-γ on the kynurenine production (Fig. 4C).

Pretreatment with IFN-γ does not alter the immunogenicity nor the immunosuppressive capacities of hMAPCs. (A) T-cells were stimulated with allogeneic irradiated (30 Gy) CD2-depleted PBMCs or hMAPCs. Human MAPCs were incubated with or without IFN-γ for 48 h prior to coculture with T-cells. After 6 days [3H]thymidine incorporation was measured. Results are expressed as mean stimulation index ± SEM of three independent experiments. Statistical significance was tested with unpaired t test. (B) Human MAPCs were added at S/R of 1:2, either with or without preincubation with IFN-γ for 48 h to an MLR. Results are expressed as mean percentage proliferation of two independent experiments ± SEM. (C) Kynurenine production was measured in culture supernatants as a consequence of indoleamine 2,3-dioxygenase (IDO) activity of hMAPCs cultured for 3 days in medium supplemented with tryptophan in the presence of varying cytokines. Left and right graph represent two different hMAPC isolations. Data are presented as mean of triplicates. TNF, tumor necrosis factor.
Alloantigen-Stimulated T-Cells Are Primed in the Presence of hMAPCs
The T-cell response upon allogeneic stimulation was completely blocked when hMAPCs were added at the beginning or during a primary MLR stimulation. Several mechanisms might play a role, among them T-cell neglection, induction of T-cell anergy, or deletion of antigenspecific activated T-cells. In a first series of experiments, rIL-2 was added to the cultures. The addition of 10 U/ml rIL-2 could not rescue the T-cell response in these experimental conditions (Fig. 5A). In the same experiments, rIL-2 induced T-cell proliferation of resting T-cells alone (from 1,913 ± 1,559 cpm to 30,589 ± 27,471 cpm) and further increased T-cell proliferation in the control MLR (from 19,5741 ± 54,210 cpm to 201,890 ± 48,445 cpm). Moreover, rIL-2 used at 100 U/ml also did not rescue the T-cell proliferative response (data not shown). Nevertheless, when T-cells, suppressed during a primary MLR by hMAPCs, were restimulated with the alloantigens, they depicted a secondary immune response against the alloantigen similar to control T-cells that responded during the primary MLR (Fig. 5B), suggesting that they had been primed to exert an effector function upon restimulation (Fig. 5C).

Alloantigen-stimulated T-cells are primed in the presence of hMAPCs. (A) T-cells were cultured with allogeneic irradiated (30 Gy) PBMCs in the absence or presence of different amounts of hMAPCs with or without addition of exogenous rIL-2 (10 U/ml). Data are expressed as mean percentage proliferation of two independent experiments ± SEM. Two-way ANOVA showed no significant main effect of addition of IL-2 (p > 0.05). The main effect of S/R ratio was significant (p < 0.01). The interaction was not significant (p > 0.05). (B, C) T-cells were stimulated in the primary MLR with allogeneic irradiated PBMCs in the presence (black bars) and absence (white bars) of hMAPCs (B). After a 2-day resting period, T-cells from the primary MLR respectively cultured in the absence or presence of hMAPCs were restimulated during 4 days (C). Results are expressed as mean counts per minute (cpm) ± SEM of three independent experiments.
The Suppressive Effect of hMAPCs Is Dependent on IDO Activity
In a next series of experiments, transwell experiments were set up to determine whether the suppressive effect on T-cell proliferation was contact dependent. Therefore, responder T-cells were cocultured with irradiated allogeneic PBMCs in the lower well of a transwell system. Irradiated hMAPCs were separated via a membrane by culturing them in the upper chamber. A partial suppression of the MLR was still observed using the transwell system (Fig. 6A), suggestive for a soluble factor involved in the suppressive effect of hMAPCs in combination with a contact-dependent inhibitory mechanism. To further demonstrate the involvement of a soluble factor, supernatant from confluent hMAPCs cultures was taken and added at different ratios to a primary MLR resulting again in a suppression of T-cell proliferation (Fig. 6B). This indicates again that the suppression of hMAPCs on T-cell proliferation is, in part, based on a soluble factor produced by hMAPCs.

Mechanism of suppression. (A) A primary MLR was performed in the lower chamber of a transwell system. Human MAPCs were added at increasing number into the upper chamber of a transwell system (black bars). Cultures were compared to a MLR without transwell system (white bars). After 6 days of culture, the inserts were removed and cells in the lower chamber were pulsed with [3H] thymidine for 8 h. Data are expressed as mean percentage proliferation ± SEM of three independent experiments. Statistical analysis of classical versus transwell MLR was performed using two-way ANOVA and showed a significant main effect of culture system (p < 0.01) and of S/R ratio (p < 0.0001). The interaction was not significant (p > 0.05). (B) Supernatant of confluent hMAPC cultures was added at different concentration to a MLR. Results are expressed as mean (±SEM) percentage proliferation of triplicates of one experiment. (C) T-cells were stimulated with allogeneic irradiated PBMCs in the absence or presence of irradiated hMAPCs at S/R ratio of 1:2 without or with addition of 2 μg indomethacin, 200 μM 1-methyl-tryptophan (1-MT), or combination of both. Data are expressed as mean cpm of five independent experiments. Statistical significance was tested with paired t test, *p < 0.05, **p < 0.01.
The mechanism of suppression is mediated at least in part via a soluble factor, so we performed tests to identify the responsible factor. In the search for the involved mediator, we analyzed supernatants of cultured MAPCs. This showed that MAPCs hardly or did not produce IFN-γ (3.4 ± 7.4 pg/ml, n = 8), TNF-α (35.7 ± 41.4 pg/ml, n = 8), IL-2 (19.2 ± 18.7 pg/ml, n = 8), and IL-10 (4.4 ± 4.2 pg/ ml, n = 8) but secreted IL-6 (861.4 ± 915.6 pg/ml, n = 8) and IL-8 (1681.9 ± 1381.3 pg/ml, n = 8). Additionally, the role of important immune suppressive factors was further functionally tested. Addition of monoclonal antibodies neutralizing IL-10 and IL-10R and/or TGF-β could not consistently prevent the inhibitory effect of hMAPCs on T-cell proliferation (data not shown). The role of prostaglandin E2 (PGE2) synthesis and IDO was studied via adding indomethacin to block PGE2 or by blocking IDO activity with 1-methyl-tryptophan. This showed that blocking of PGE2 synthesis had no effect on the suppressive effect of hMAPCs on T-cell alloreactivity. On the other hand, blocking of IDO activity resulted in a slight inhibition of the T-cell proliferation, which could however not be further suppressed by hMAPCs (Fig. 6C). This suggests that the inhibitory capacity of hMAPCs is at least in part dependent on IDO activity.
Human MAPCs Are as Suppressive as hMSCs
At last, we compared the immune phenotype, immunogenicity, and immunomodulatory capacities of hMAPCs with those of its clinical grade product MultiStem and hMSCs all isolated from the same donor. Immune phenotypic characterization of the single-donor stem cell products showed that MultiStem and hMSCs are phenotypically similar to hMAPCs, except that hMSCs express significantly higher levels of MHC class I (Table 2). To compare the immunogenicity of single-donor hMAPCs, MultiStem, and hMSCs, purified responder T-cells were stimulated with all three irradiated, allogeneic single-donor stem cell products and CD2-depleted PBMCs as a positive control. Stimulator cells were cocultured with T-cells at a responder/stimulator ratio of 2:1. After 6 days, T-cell proliferation was measured by means of [3H]thymidine incorporation. This showed that MHC identical hMAPCs, MultiStem, and hMSCs were all similarly low immunogenic in vitro (Fig. 7A). In addition, the immunosuppressive effect of hMAPCs was then compared to those induced by MultiStem and hMSCs derived from the same donor. This revealed that hMAPCs and MultiStem were as suppressive as hMSCs (Fig. 7B).
Comparison of the Immune Phenotype of hMAPCs, hMSCs, and MultiStem
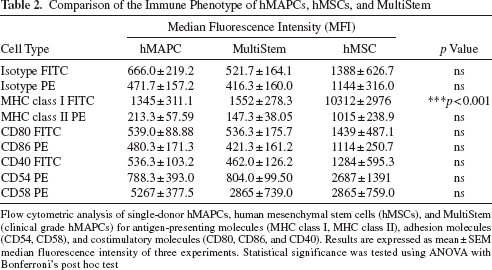
Flow cytometric analysis of single-donor hMAPCs, human mesenchymal stem cells (hMSCs), and MultiStem (clinical grade hMAPCs) for antigen-presenting molecules (MHC class I, MHC class II), adhesion molecules (CD54, CD58), and costimulatory molecules (CD80, CD86, and CD40). Results are expressed as mean ± SEM median fluorescence intensity of three experiments. Statistical significance was tested using ANOVA with Bonferroni's post hoc test

Human MAPCs are as immunosuppressive as hMSCs. (A) T-cells proliferation upon stimulation with allogeneic irradiated (30 Gy) single-donor CD2-depleted PBMCs, hMAPCs, MultiStem, and hMSCs. Results are expressed as mean stimulation index ± SEM (n = 5). Statistical significance (**p < 0.01) compared to control with CD2-depleted PBMC was tested with ANOVA and Bonferroni's post hoc test. The comparison of hMAPCs to hMSCs was not statistically significant. (B) T-cells were stimulated with allogeneic irradiated PBMCs in the absence or presence of different doses of irradiated single-donor hMAPCs, MultiStem, or hMSCs. Data are expressed as mean (±SEM) percentage proliferation as compared to control MLR in the absence of stem cells. Results are representative of three independent experiments. Two-way ANOVA showed no significant main effect of the suppressor cell type (p > 0.05). The main effect of S/R ratio was significant (p < 0.0001). The interaction was not significant (p > 0.05).
Discussion
We herein describe for the first time the immunological properties of hMAPCs. Recently, it has been published that hMAPCs display different phenotypical and functional characteristics as compared to hMSCs (25), which made the study of the immunological characteristics of hMAPCs inevitable. We now demonstrate that hMAPCs are nonimmunogenic and exert strong immunosuppressive effects on T-cell responses in vitro.
Human MAPCs do not induce proliferation of allogeneic T-cells, and this is consistent with their immune phenotypic characteristics, such as the lack of MHC class II and costimulatory molecules belonging to the B7 family. Pretreatment with IFN-γ, as a model of an inflammatory environment in which cells might be delivered in vivo, led to an upregulation of MHC class I, MHC class II, and ICAM-I. However, IFN-γ pretreatment did not enhance immunogenicity of MAPCs, and this might be related to the concomitant increase of IDO expression. Human MAPCs also do not induce production of Th1 or Th2 cytokines upon coculture with allogeneic T-cells, although low levels of activation markers on responders T-cells are induced. In addition, we showed with multiple samples used in multiple experiments that hMAPCs have suppressive effects on allogeneic T-cell proliferation. The suppressive effect of hMAPCs is very consistent, as we observed similar suppression on T-cell alloreactivity with hMAPCs from all different donors. The suppressive effect was not influenced by MHC compatibility as both autologous and third-party stem cells show similar suppression. This is of utmost importance because hMAPCs will be used as an off-the-shelf stem cell product for cellular therapy. Human MAPCs also suppress T-cell proliferation of memory T-cells upon stimulation with recall antigens and of effector T-cells during a secondary MLR. Delayed addition of hMAPCs to an MLR showed the same inhibitory effect. These data suggest that hMAPCs can also suppress an ongoing immune response. This finding is of importance when hMAPCs will be used in clinical settings not only for prevention but also for treatment of immune-mediated diseases. Finally, the suppressive effect of hMAPCs is not influenced by pretreatment with IFN-γ. Hence, hMAPCs might remain immunosuppressive when injected into an inflammatory environment in vivo. In general, our data are in line with published data on the immunological properties of hMSCs. We confirmed this similarity by isolating hMAPCs, its clinical grade stem cell product MultiStem, and hMSCs from two single donors. Both the immunogenicity and immunosuppressive capacities were comparable between all three stem cell products. This indicates that hMAPCs with their potency of large-scale expansion are a valuable alternative source of immunomodulatory cellular stem cell product.
The mechanism responsible for the immunosuppression of MAPCs is not yet completely understood. Data on hMSCs are often contradictory because of different experimental designs to study immunosuppression. First, we showed that, although hMAPCs strongly suppress T-cell allogeneic responses during a primary MLR, they do not prevent priming of T-cells, as suppressed T-cells show similar secondary responses. This indicates that hMAPCs do not induce T-cell anergy. Di Nicola et al. (4) reported that the suppressive effect of hMSCs was reversible, but in their study T-cells were stimulated with a mitogen during the primary MLR instead of allogeneic PBMC as in our experiments. Second, transwell experiments demonstrated that hMAPCs still exert a suppressive effect, albeit lower, when they are separated from the effector T-cells via a membrane. This suggests that the suppression is mediated, at least in part, via a soluble factor which blocks T-cell proliferation. This hypothesis is supported by the immunosuppressive activity of added supernatants. However, similar as shown for hMSCs (4, 28), the degree of suppression was stronger when cell–cell contact was present. This points to a parallel cell contact-dependent suppressive activity or to adhesion of immune cells to MAPCs, which strengthen their suppressive function through soluble factors. Several candidate molecules have been proposed as the soluble immunosuppressive factor produced by hMSCs, although data are contradictory. Antibodies against TGF-β have been shown to partially restore proliferation of purified T-cells stimulated with allogeneic peripheral blood lymphocytes (4), but this was not involved in the suppressive effect when T-cells were stimulated with mitogens (13) or when PBMCs were used as responder cells (22, 28). Aggarwal et al. (1) reported that inhibition of the synthesis of PGE2 mitigated MSC-mediated suppression. This was confirmed by Rasmusson et al. (23), who saw a restoration of T-cell proliferation suppressed by hMSCs with indomethacin, but only when T-cells were stimulated with PHA, and not in a MLR. Although tryptophan depletion was not responsible for an immunosuppressive effect of hMSCs when unfractionated PBMCs were used as responder cells (28), Meisel et al. (20) could demonstrate a role for the tryptophan catabolizing enzyme IDO in the suppression by hMSCs, and that expression of IDO was induced by IFN-γ. These two observations are similar to what we observed. Human MAPCs display increased IDO activity upon IFN-γ pretreatment and lose their suppressive effect on T-cell proliferation by blocking IDO activity. The data confirm that the suppressive effect of hMAPCs is at least in part dependent on IDO activity.
The finding that the immunological characteristics of hMAPCs are similar to those of hMSCs was encouraging to move forward and start a phase one trial for the prevention of GVHD. One major advantage of using hMAPCs instead of hMSCs is the more pronounced expansion potential, making it possible to use one uniform lot of stem cells to perform a complete clinical trial. This allows more accurate interpretation of the results from the clinical trial bypassing conflicting data due to the use of a heterogeneous stem cell population. In addition, our data pave the way to use hMAPCs as an off-the-shelf stem cell product not only to prevent an alloresponse but also to treat ongoing immune responses. Indeed, the immunomodulation by hMAPCs was MHC independent and could similarly suppress an ongoing immune response. The use of a third-party off-the-shelf stem product permits immediate administration of the product when needed.
Until now, data on the immunological properties of MAPCs have only been published for cells derived from mouse and rat bone marrow. Murine MAPCs, like hMAPCs, were nonimmunogenic for T-cells and were capable to suppress an allogeneic T-cell response (6, 18, 26). However, in contrast to the hMAPCs, the mechanism of suppression was mediated by PGE2 alone (6). Rat MAPCs have been reported to suppress alloreactive T-cell proliferation via IDO (9). These differences in mechanism of suppression between mouse, rat, and human are also seen for MSCs (24). The immunosuppressive mechanism of mMSCs in alloimmune responses have been described recently by Han et al. (5). Documented in vitro differences between species mean that it will be difficult to extrapolate the results of in vivo studies on the immunomodulatory capacities of stem cells into relevant clinical information (19, 24). Different experimental animal studies have been used to assess the immunomodulatory properties of MSCs in vivo (21). As for the MAPCs, Luyckx et al. proposed the popliteal lymph node assay to demonstrate in vivo the immunomodulatory effect of murine MAPCs (17). The same model can be used to study the immunomodulatory function of hMAPCs.
We conclude that hMAPCs have immunological properties similar to those from hMSCs, at least in vitro. The role of hMAPCs as immunomodulatory cellular stem cell products in vivo is the focus of current clinical research. Our data support the currently running phase 1 clinical trials with MultiStem for the prevention of GVHD, cardiac ischemia, and Crohn's disease. Human MAPCs as well as MultiStem have significantly greater expansion potential with stable genetics, compared with hMSCs. This allows the creation of master cell banks to derive large number of cell lots for clinical use. Standardized quality control of such advanced therapy medicinal products aimed for immunomodulation includes standardized immunosuppressive potency tests for each batch. The standardization and the rationale of the clinical application are strongly supported by the data provided in this study.
Footnotes
Acknowledgments
The authors would like to thank Lieve Coorevits, Kathleen De Swert, Elke Nackers, and Anaïs Van Hoylandt, Catholic University of Leuven, for their excellent technical assistance. We would also wish to thank Vik Van Duppen, Catholic University of Leuven, for his help with the flow cytometry and Ahmad Kasran, Catholic University of Leuven, for the fruitful discussions, technical assistance, and his help with the statistical analysis. We are thankful to Walter Däubener, Heinrich-Heine-University of Düsseldorf, for the measurement of Kynurenine. This work was supported by the Center of Excellence (EF/05/011) funding Catholic University of Leuven, an Odysseus award, research funding from Athersys, Inc., and a grant from the European Commission (EC-FP6-STREP-STROKEMAP) to Catherine M. Verfaillie. Valerie D. Roobrouck and Ariane Luyckx are funded by a grant from the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT Vlaanderen). Stefaan W. Van Gool is senior clinical investigator of FWO Vlaanderen. Catherine M. Verfaillie is a consultant to Athersys, Inc., and research funds were obtained from Athersys, Inc. Jef Pinxteren is an employee of ReGenesys, and Robert Deans and Wouter van't Hof are employees of Athersys, Inc.