
Abstract
Recombinant proteins are an important tool for research and therapeutic applications. Therapeutic proteins have been delivered to several cell types and tissues and might be used to improve the outcome of the cell transplantation. Recombinant proteins are propagated in bacteria, which will contaminate them with the lypopolysacharide endotoxin found in the outer bacterial membrane. Endotoxin could interfere with in vitro biological assays and is the major pathological factor, which must be removed or inactivated before in vivo administration. Here we describe a one-step protocol in which the endotoxin activity on recombinant proteins is remarkably reduced by transient exposure to acidic conditions. Maximum endotoxin deactivation occurs at acidic pH below their respective isoelectric point (pI). This method does not require additional protein purification or separation of the protein from the endotoxin fraction. The endotoxin level was measured both in vitro and in vivo. For in vitro assessment we have utilized Limulus Amebocyte Lysate method for in vivo the pyrogenic test. We have tested the above-mentioned method with five different recombinant proteins, including a monoclonal antibody clone 5c8 against CD154 produced by hybridomas. More than 99% of endotoxin was deactivated in all of the proteins; the recovery of the protein after deactivation varied between maximum 72.9% and minimum 46.8%. The anti-CD154 clone 5c8 activity remained unchanged as verified by the measurement of binding capability to activated lymphocytes. Furthermore, the effectiveness of this method was not significantly altered by urea, commonly used in protein purification. This procedure provides a simple and cost-efficient way to reduce the endotoxin activity in antibodies and recombinant proteins.
Introduction
Many recombinant proteins with commercial and medical applications are produced utilizing genetically modified bacteria (E. coli), an expression system characterized by a quick production of highly concentrated protein. The decontamination from endotoxin during the purification process is a prerequisite for most recombinant proteins used either in vitro or in vivo.
Endotoxin is a lipopolysaccharide (LPS) forming an integral part of the Gram-negative bacteria outer membrane responsible for bacterial organization and stability (47). In vivo exposure to endotoxin-contaminated products triggers adverse reactions such as rising body temperature, activation of coagulation cascade, modification of hemodynamics, and septic shock (26). Endotoxin can significantly modify interpretation of in vitro experiments (11). Therefore, its removal or deactivation is a critical step in production of E. coli-generated recombinant proteins.
Because endotoxin is temperature resistant, its elimination is one of the most difficult steps in the protein purification process (42). The commonly used methods are the following. 1) The anion exchange chromatography (25) that utilizes the negative net charge of endotoxin and its binding to the anion exchange resin. This method can be applied only to basic proteins that do not bind to anion exchange columns (2,6,25,34). 2) The use of adsorbents such as histidine, histamine, polymyxin B, and poly-l-lysine to facilitate adsorption of endotoxin to matrix by electrostatic and hydrophobic interactions. Even though the interaction between adsorbent and endotoxin is selective, the multiple rounds of the treatment and repeated protein dilutions decrease product recovery (3). 3) The use of Triton X-114, which is an efficient compound for endotoxin removal from recombinant proteins (1,23), and it can be used either in a phase separation method (23) or in washing solutions for affinity chromatography columns (37). However, traces of the detergent must be removed by repeated adsorptions or gel filtrations, which again may decrease recovery of the purified protein. This study reports an additional method for bacterial endotoxin deactivation on recombinant proteins. The goal is achieved by acidic pH treatment. Interestingly, we found that this technique does not require further protein purification or separation from endotoxin fraction. We have achieved up to 99.9% reduction of endotoxin activity in freshly purified recombinant protein, with protein recovery of 72.9% as assessed by colorimetric protein assay from BioRad. Moreover, we have successfully applied this technique to deactivate endotoxin in monoclonal antibodies produced by hybridomas.
Materials and Methods
Protein Generation and Purification
Small frozen stock aliquot of E. coli (BL21) transformed with plasmid coding for protein of interest was inoculated into 100 ml of LB/Amp medium supplemented with antibiotics and incubated for 5 h at 37°C with continuous agitation. The 100-ml culture was transferred to 1 L of LB/Amp media and grown for additional 16 h under the same conditions. The resulting bacterial culture was centrifuged; the pellet was washed once with PBS and resuspended in 30 ml of PBS supplemented with protease inhibitors (Complete, EDTA free; Roche) and 20 mM imidazole. The suspension was sonicated in three repetitions of 21 s, 50% amplitude, and 1-min cool off period (Fisher Scientific Sonic Dismembrator, Model 500). The resulting bacterial extract was centrifuged 45 min at 12,000 rpm and the supernatant was loaded on Niagarose column (Qiagen) previously equilibrated with 20 mM imidazole buffer. The protein bound to the column was washed with 20 and 30 mM imidazole buffers and eluted with 100 mM imidazole buffer. The eluted protein was desalted on PD-10 column (Amersham-Pharmacia). Fractions were monitored with colorimetric protein assay kit (Bio-Rad Cat. #500-0006) and quantified by spectrophotometer (Beckman-DU 640) at 595 nm wavelength.
For in vivo experiments, the protein solution was sterilized by passage through the 0.2-μm syringe filters (Acrodisc HT Tuffryn Membrane Low Protein Binding Non-pyrogenic).
Recombinant proteins used in this study were: murine heme oxigenase 1 (HO-1), cell-permeable mouse TAT heme oxigenase 1 (TAT-HO-1) (37), human neuroglobin (Ngb) (27), cell-permeable human TAT-transcription factor PDX-1 (TAT PDX-1), and β-galactosidase (41). To one of the preparations of TAT-HO-1, 6 M urea solution was added to investigate if urea has a negative effect on the described endotoxin decontamination method. HO-1 and TAT-HO-1 proteins were chosen for further experimentation because they are relatively easy to purify in large quantities.
Endotoxin Quantification
The level of active endotoxin was quantified using one of two Limulus Amebocyte Lysate (LAL) methods: 1) QCL 1000 kit from Cambrex/Biowhittaker (Cat. #50–647U) with Fluorescent Plate reader FL600 at 405 nm wavelength using Bio-Tek (USA) KC4 software; 2) Endosafe PTS (Cat. #PTS 100)-Charles Rivers Laboratories with cartridge sensitivity between 10 and 0.1 (Cat. #PTS 201) or 5 and 0.05 EU/ml (Cat. #PTS 2005). All dilutions tested were properly spiked to confirm results.
Endotoxin Deactivation
The pH of the recombinant protein solution or monoclonal antibody clone 5c8 was adjusted to acidic conditions as follows: the protein was purified as described above; the concentration was corrected to 0.5 mg/ml and assessed utilizing colorimetric Protein-Assay (Bio-Rad). The pH of 5 ml protein solution was adjusted dropwise with constant stirring using 1 M HCl and monitored with pH meter. After the protein reached desired acidic pH it was incubated at room temperature for 20 min. At the end of incubation the pH was slowly returned to its original value with 1 M NaOH and the protein solution was filtered by passage through the 0.2-μm Acrodisc syringe filter. The concentration of the recovered protein was measured by colorimetric protein assay kit (BioRad) and the protein was subjected to functional studies. 5c8, the hybridoma clone against CD154 (CD40 ligand), was subjected to the above-described procedure twice at pH 5. All tubes were purchased endotoxin free (Corning, Falcon conical tubes or borosilicate pyrogen-free test tubes; Lonza, Cat. #N207). The pH electrode was pre-soaked in 0.1 M HCl and washed extensively with pyrogene-free water before each use.
Assessment of Endotoxin Activity In Vivo
Pyrogenic tests were performed with two groups of three male rabbits each, weighing between 2.2 and 3.2 kg. The rectal probe was inserted and each animal was allowed to acclimate to its restrained position for 1 h. Four temperature readings at 30-min intervals were recorded and the average of all four readings was registered as a base line temperature. The animals were than infused for 4 min via the marginal ear vein with pre-warmed protein solution (37°C) and appropriate volume per kilogram (3.5–4 ml/kg) with no significant differences in solution volume between groups. The first group (group 1) received solution with TAT-HO-1 protein (38) that underwent endotoxin deactivation at pH 5 with the final endotoxin level of 0.3 EU/ml measured by LAL. The second group (group 2) received the TAT- HO-1 preparation that was not subjected to endotoxin treatment and contained 20 EU/ml, similar amounts of protein were injected in both groups. Prior to infusion the samples were equally diluted. The dilutions were made to prevent occurrence of the lethal pyrogenicity in the nontreated group (group 2). All pyrogenic in vivo testing was done by NAMSA (Irvine, CA, USA), a fully accredited company that performs in vivo pyrogen assessments. After the infusion the temperatures were recorded at 30-min intervals for 3 h. All procedures were conducted in compliance with good laboratory practice, SO 17025 and met nonpyrogenic requirements for United States Pharmacopeia (USP).
Peripheral Blood Lymphocytes Activation
Peripheral blood lymphocytes (PBL) from the freshly drawn blood of healthy cynomolgus monkey (Mann-heimer, Miami FL) were isolated on Ficoll-Hypaque (Sigma Chemical Co., St. Louis, MO) following manu-facturer' instructions. The purified PBLs (10 × 106/ml) were cultured at 37°C in a humidified atmosphere of 5% CO2 for 6 h in RPMI-1640 medium (Gibco-BRL, NY) supplemented with 10 ng/ml of PMA (Sigma Chemical Co.), 10 mg/ml of PHA (Sigma Chemical Co.), 10% heat-inactivated FBS, 100 U/ml penicillin, 100 mg/ml streptomycin, 10 nM sodium pyruvate, 2 mM l-glutamine, 0.1 mM nonessential amino acids, and 1x vitamins (all from Gibco-BRL, NY). After T-cell activation, cells were washed twice with PBS (Gibco-BRL, NY) and immunostained.
The 5c8 is a protein A purified monoclonal antibody that was specially generated by an original 5C8 hybridoma cell line obtained from ATCC (Cat. #HB-10916, Lot #222225) through Rockland (www.rocklandinc.com) with Code #CUST22M and Lot #17544C.Clone 5c8 anti-CD154 is being used extensively in vivo and in vitro for CD40 signaling research at the Diabetes Research Institute and therefore was a logical choice for our experiments.
Statistical Analysis
Results from protein acidification at pH 5 and 6 were analyzed using Wilcoxon signed rank test for paired, nonparametric samples, with 95% confidence intervals. Endotoxin activity and binding efficiency of anti-CD154 before and after acidification were analyzed using two-tailed Student t-test with a value of p < 0.05 considered statistically significant.
Results
Endotoxin Activity Decreases After Acidic pH Treatment
Initially, we have observed that the endotoxin activity in recombinant proteins was dramatically reduced by lowering the pH of the protein to acidic range. We have verified this method with a number of recombinant proteins, each with a different isoelectric point (pI). The experiments were performed with a variety of pH values (Table 1). Overall, we detected a decrease in endotoxin activity at acidic pH below the respective protein pI. The endotoxin activity was not significantly decreased at the basic pH range (data not shown). The subsequent protein recovery seems to depend on the protein type. For example, even though more than 99% of endotoxin was deactivated in HO-1, TAT-HO-1, Ngb, and TAT-PDX the recovery of each protein varied from 90.4%, 72.9%, 60.8%, to 46.8%, respectively. We have compared effectiveness of endotoxin deactivation under acidic pH conditions using either plastic containers (Corning or Falcon conical) or borosilicate pyrogen-free test tubes. As long as the acidic conditions were upheld the material of which the vessel was made had no effect on the final results (data not shown). At neutral pH, proteins in plastic containers showed slow endotoxin decrease over time (Tables 1 and 2).
Endotoxin Activity of Protein Solutions at Different pH

pI was calculated using Swiss-Prot tools (http://www.expasy.org).
Effect of pH Changes on Endotoxin Activity and Protein Recovery of TAT-HO1

Values are mean ± SE.
p = 0.05 from Wilcoxon two sample test.
Once the preliminary screening of various proteins was completed, we studied the reproducibility of this procedure and purified 10 TAT-HO-1 preparations at pH 5 and 12 preparations at pH 6 (Table 2). Endotoxin was efficiently deactivated at pH 5; the remaining activity was negligible at 0.31 ± 0.11% EU/mg (n=10; p = 0.00018). On average, the protein recovery was 64.45 ± 3.87%. The endotoxin activity varies greatly from preparation to preparation. The treated TAT-HO-1 did not lose its biological activity, assessed as the capability to protect the insulinoma cells βTC3 from death induced by a combination of 10 μg/ml cyclohexamide and 4000 U/ml TNF-α (38). There was virtually no difference in function of treated and untreated protein (data not shown).
Efficient Endotoxin Deactivation in the Presence of Urea
Because urea is frequently used in recombinant protein purification protocols, we tested if it has adverse effect on our method of endotoxin deactivation. In urea-free recombinant protein, the endotoxin activity was reduced from 8107.55 to 5.57 EU/mg (99.9% activity reduction); in the presence of 6 M urea the final concentration of remaining activity was 49.62 EU/mg (99.4% activity reduction). The protein recovery was similar for both conditions 70.72 ± 7.48% and 73.71 ± 8.27%, respectively.
In Vivo Pyrogenic Determination
The in vivo pyrogen assay with rabbits confirmed the results obtained with LAL in vitro assays in regards to endotoxin deactivation. Figure 1 demonstrates untreated TAT-HO1 protein with high endotoxin activity as fully pyrogenic while the treated TAT-HO1 protein with very low endotoxin activity shows no pyrogenic response, which correlates the in vivo studies with the in vitro LAL based results.
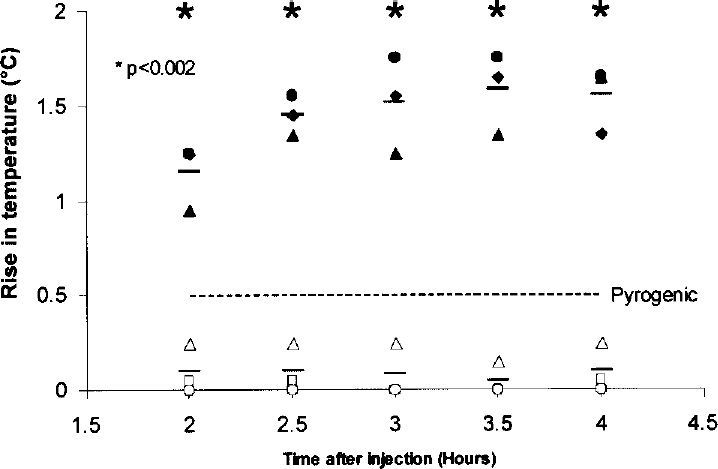
In vivo determination of pyrogenicity of TAT-HO1 protein following endotoxin deactivation. Pyrogenicity of TAT-HO1 protein (solid symbols) or TAT-HO-1 protein after endotoxin decontamination (open symbols) was tested in vivo using male rabbits (n = 3 per group). Animals were injected intravenously (marginal ear vein) with 3.5–4 ml/kg of the indicated protein. The group receiving endotoxin-contaminated TAT-HO1 protein (solid symbols) showed pyrogenic reaction higher than 0.5°C above baseline temperature, while the group of animals receiving the protein that had undergone endotoxin decontamination had no pyrogenic reaction. Data are an average of three animals per group. The broken line indicates the cut-off for pyrogenicity (namely, 0.5°C). Unpaired t-test: ∗p < 0.002 at each time point.
Effect of Endotoxin Deactivation on Monoclonal Antibody Produced by Hybridoma
Low levels of endotoxin activity (0.51 EU/mg; Table 3) were found in proteins produced in sources other than bacteria, such as monoclonal antibody produced by hybridoma clone 5c8 (ATCC, Manassas, VA; Cat. #HB-10916). The monoclonal antibody 5c8 is a clone directed against human CD154 (CD40 ligand) (20). After two rounds of acidic treatment at pH 5 the activity levels of endotoxin were no longer detectible by our method of assessment. The loss of protein was minimal (Table 3). Activity of the antibody measured by the binding capability of the 5c8 to in vitro-activated PBLs expressing CD154 showed no significant difference between treated and untreated samples (Table 3, Fig. 2).

Activity/function of antibody 5c8 was measured by flow cytometry with (A) pre- or (B) postendotoxin deactivated samples (n = 3).
Endotoxin Activity in 5c8 Antibody (Anti-CD154) and Binding Efficiency of the Protein Before and After Treatment

bd: below detection.
p = 0.95 t-test two tailed, p-value nonsignificant.
Discussion
With the progress of genomics and proteomics in recent years, the production of recombinant therapeutic proteins and peptides has increased significantly. Therapeutic proteins and peptides have been delivered to several cell types or tissues such as brain (19,29,32,36), muscle (5,8,49,50), heart (3,15), eye lens epithelial cells (28), and into living organisms (43). The recombinant proteins are considered for use in cell transplantation as well (22,33). Furthermore, human cells were successfully transduced with artificial transcriptional regulatory proteins, such as zinc-finger transcription factors (35). Recombinant proteins with antiapoptotic/necrotic properties were delivered to islets postisolation and prior to transplantation to protect them from stress. The transduction reduced apoptosis and necrosis of transplanted islets (13,33). Recent reports indicate that delivery of recombinant transcription factor PDX-1 has a potential to treat diabetes (21). Recombinant proteins have been used to engineer stem cells or progenitor cells in order to differentiate them into cell types such as insulin producing cells (β-cells) (10,31,46,51), hematopoietic cells (24), and neurons (17,44), which could be used to treat diabetes, leukemia, and diseases of nerves system, respectively. Recombinant proteins have been used to re-program induced pluripotent stem cells as well (4,52). Most of these proteins are produced in bacteria and endotoxin deactivation should be a mandatory step of the purification.
We have found that after short incubation at acidic pH, the endotoxin activity of most recombinant proteins has been dramatically reduced as assessed by in vitro and in vivo assays. We applied the method to different recombinant proteins. In order to optimize efficiency of endotoxin deactivation and protein recovery, it was necessary to test treatments with various acidic pH for each target protein (Table 1). Optimal endotoxin deactivation was observed at pH below the isoelectric point of the treated protein (Table 1).
The finding that endotoxin activity may be dramatically reduced in protein preparations merely by acidic pH treatment, without any further processing, was surprising. Biologically active endotoxin, in the form of mi-cellar aggregates (28), is capable of interacting with multiple proteins forming complexes based on electrostatic and hydrophobic interactions (25,34). Due to these interactions the endotoxin removal or deactivation from the protein preparations was one of the most challenging steps of the purification. Dissociation of the protein–endotoxin complex is considered a prerequisite for endotoxin removal by biochemical procedures (25). Several reports described acidic pH treatment prior to purification such as affinity chromatography (16,39), affinity adsorption (9,48), ion exchange resin (30), and filtration (14,18).
The decrease in pH is believed to aid the dissociation of endotoxin–protein aggregates for subsequent separation using the methods described above. However, our studies show that endotoxin is no longer active after acidic pH treatment and therefore no further removal is required. Interestingly, examples of endotoxin conformations that are inactive have been previously described (12,40,45). Conformation changes of endotoxin aggregates are also known to occur at different pH ranges (7). Therefore, we speculate that the acidic treatment irreversibly changes the endotoxin conformation, which causes the loss of its activity in vitro and in vivo. Further biophysical and biochemical studies will be necessary to shed the light on the mechanism responsible for the inhibition of endotoxin activity after acidic treatment.
The method we have described here is not exempt from limitations. It requires optimum pH screening, for maximum protein recovery. After endotoxin decontamination, the protein should be tested for chemical integrity and biological activity. In some cases the endotoxin deactivation as described above was not effective. For example, we tested the method on commercially available collagenase prepared from gram-positive Clostridium histolyticum (Sigma-Aldrich Cat. #C9263) that had low levels of endotoxin (~70 EU/mg) contamination. Exposure to acidic conditions induced irreversible precipitation of collagenase, rendering the method inapplicable for this particular protein. In contrast, it successfully deactivated the endotoxin in 5c8 antibodies produced in hybridoma cells (Fig. 2). Small amounts of endotoxin contamination required two rounds of acidic pH treatment in order to enhance the efficiency of the method, which brought the endotoxin activity below the detection levels (Table 3).
In conclusion, this study describes a simple and inexpensive procedure to reduce endotoxin activity in proteins able to withstand acidic pH treatment. Because the previous endotoxin elimination technique is a complicated step in protein purification it could be worth to test our simple method before embarking on more complex procedures.
Footnotes
Acknowledgments
This work was supported by a grant from the National Institutes of Health (DK-59993 to R.L.P.). We thank Dr. Antonello Pileggi for critical review of this manuscript.