
Abstract
BACKGROUND:
Shanmei Capsule is a famous preparation in China. However, the related mechanism of Shanmei Capsule against hyperlipidemia has yet to be revealed.
OBJECTIVE:
To elucidate underlying mechanism of Shanmei Capsule against hyperlipidemia through network pharmacology approach and molecular docking.
METHODS:
Active ingredients, targets of Shanmei Capsule as well as targets for hyperlipidemia were screened based on database. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment were performed via Database for Annotation, Visualization, and Integrated Discovery (DAVID) 6.8 database. Ingredient-target-disease-pathway network was visualized utilizing Cytoscape software and molecular docking was performed by Autodock Vina.
RESULTS:
Seventeen active ingredients in Shanmei Capsule were screened out with a closely connection with 34 hyperlipidemia-related targets. GO analysis revealed 40 biological processes, 5 cellular components and 29 molecular functions. A total of 15 signal pathways were enriched by KEGG pathway enrichment analysis. The docking results indicated that the binding activities of key ingredients for PPAR-
CONCLUSIONS:
The possible molecular mechanism mainly involved PPAR signaling pathway, Bile secretion and TNF signaling pathway via acting on MAPK8, PPAR

Introduction
Hyperlipidemia is one of the most prevalent global chronic metabolic diseases, which is closely related to many high-incidence cardiovascular diseases, such as hypertension, diabetes, coronary heart disease and atherosclerosis [1, 2, 3]. It is characterized as elevated contents of total cholesterol (TC), triglyceride (TG), low-density-lipoprotein cholesterol (LDL-C), as well as decreased high-density-lipoprotein cholesterol (HDL-C) levels [4]. With the elevating prevalence and morbidity, hyperlipidemia has become a critical global issue for public health. Regulating lipid metabolism disorders may be a potential approach to decelerating or preventing the progression of hyperlipidemia. At present, statins, such as atorvastatin, lovastatin and simvastatin, are among the most commonly used lipid-lowering drugs to reduce plasma lipids. Statins are generally regarded as safe and tolerable drugs, however, certain controversies remain. A survey analyzed all case reports from the FDA AE Reporting System database linking muscle-related adverse events to statin use. The results show that dose-dependent side effects occur across the statin class [5, 6, 7, 8]. Thus, it is necessary to develop and identify an alternative drug that may be valuable in regulating lipid metabolism. Compound formulae of traditional Chinese medicine (TCM) emphasizes emphasizes the synergism among active ingredients, with proper herbs and relevant dosage to synergize the desirable effects and minimize side effects integrally. Compared with Western medicine, TCM are considered relatively safe and produces fewer adverse reactions [9, 10].
TCM has been used for thousands of years for the treatment of hyperlipidemia in China, with advantages of multitarget mechanisms, remarkable curative effect and safety. They are featured by global regulation of glucose and lipid metabolism, as well as energy homeostasis via active ingredients in a prescription [11]. Shanmei Capsule, a famous preparation in China, is made of a combination of Crataegi Folium (called Shanzhaye in Chinese, the leaves of Grataegus pinnatifida Bge. var. major N.E. Br. or Grataegus pinnatifida Bge.) and Dahurian rose fruit (called Cimeiguo in Chinese, the fruit of Rosa davurica Pall. var. davurica) ethanol extract in proportion [12]. Crataegi Folium and Dahurian rose fruit both are well-known traditional medicinal plants, which have been confirmed to possess various biological and pharmacological activities, such as anti-inflammatory, antioxidant, hypocholesterolaemic and hypolipidaemic effects [13, 14, 15]. In clinical application, they are included in remedies on their own or in combination with other herbs to regulate lipid metabolism disorders induced by cardiovascular diseases. The above evidence suggests that Shanmei Capsule may be an alternative approach for treating hyperlipidemia. However, the hypolipidemic effect and related activity mechanisms of Shanmei Capsule against hyperlipidemia have yet to be revealed. Due to the complexity of active ingredients and the unknown synergistic effects of multiple components, it remains a laborious task to clarify the molecular mechanism of the Shanmei Capsule.
With the booming development of systems biology and bioinformatics, network pharmacology approach has been used successfully for the discovery of the key targets and their potential molecular mechanisms [16, 17]. Contrary to “one drug, one target, one disease” principle of traditional drug design, network pharmacology based on the “drug-disease-target” interaction network, which comprehensively and systematically evaluates the intervention or effect of drugs on diseases. Network pharmacology combines the multi-level information of “ingredient-disease-target-pathway”, which corresponded with the characteristics of TCM. Molecular docking is an important computer-aided drug discovery method, which can be used to predict the interactions between drug ingredients and target proteins, and the possible binding sites of drugs [18, 19]. Interactions between drug molecules and target proteins provide important information on drug discovery, which will help towards the development of drugs to treat hyperlipidemia.
In this study, network pharmacology approach was applied to investigate the relationships between active ingredients, core targets and pathways. Based on the results of network analysis and molecular docking, multi-ingredient, multi-target, and multi-pathway potential mechanisms related to Shanmei Capsule against hyperlipidemia were predicted.
Methods
Collection of active ingredients for Shanmei Capsule
Crataegi Folium and Dahurian rose fruit were used as keywords to screen the candidate ingredients of Shanmei Capsule. All the chemical ingredients were queried based on the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP) (http://tcmspw.com/tcmsp.php) and literatures. The structural information regarding biological activities of small molecules, including CAS numbers and Canonical SMILES strings, were obtained from the Scifinder database (https://scifinder.cas.org/scifinder/) and PubChem database (https://pubchem.ncbi.nlm.nih.gov/) [20].
Target acquisition for drug and disease-related
Target prediction of Shanmei Capsule was performed using Swiss Target Prediction (http://www.swisstargetprediction.ch/) [21]. The Canonical SMILES strings obtained in 2.1 was introduced into the Swiss Target Prediction to obtain human-related targets for active ingredients. Using hyperlipidemia as a keyword, the candidate target of hyperlipidemia was searched by the GeneCard database (version 4.14, https://www.genecards.org/). After removing the duplicate targets, the overlapping targets related to active ingredients and hyperlipidemia were selected as the candidate targets, and then imported into the STRING database (http://string-db.org/) for gene interaction analysis.
GO and KEGG pathway enrichment analysis
Gene Ontology (GO) functional enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway enrichment analysis were carried out based on the Database for Annotation, Visualization and Integrated Discovery (DAVID, version 6.8, http://www.david.niaid.nih.gov). Results with values of
Network construction and analysis
Network construction and analysis were performed using Cytoscape 3.3.0 (download from https://cytoscape.org/). The nodes in different shapes and colors of the network represented active ingredients, targets, pathways, and the edges indicated the interaction of them. In this study, degree refers to the number of directly connected nodes with one another. It is considered that the greater the number of degree is, the greater the influence is. The topological parameters of network, such as degree, were calculated by Network Analyzer (Cytoscape plugin).
Molecular docking
The candidate ingredients of Shanmei Capsule and lifibrate (positive control drug) were chosen for molecular docking, and the results were visualized using PyMOL software (https://www.pymol.org/). The detailed steps were as follows. Firstly, molecular structure of active ingredients and the protein crystal structure of the hyperlipidemia-related target were downloaded from the ZINC 15 (http://zinc.docking.org/) and RCSB PDB database (https://www.rcsb.org/), respectively. Prior to performing the docking process, the target protein structure was pretreated by AutoTools, including removal of water molecules, addition with hydrogens and so on. Secondly, the active pocket on protein structure was predicted by GetBox Plugin (PyMOL plugin) [22, 23]. The values of the utilized parameters in autodock software were presented in Table S1. Finally, molecular docking simulation was performed using the Autodock Vina software. According to the scoring method, the docking models with the lowest score and binding-energy value were selected and visualized for subsequent analysis.
ADME and toxicology prediction
The 2D structures (in .sdf format) and simplified molecular input line entry specification (SMILES) codes of 17 key active ingredients were downloaded from the PubChem database (http://pubchem.ncbi.nlm.nih.gov). Then, a SwissADME web tool (http://www.swissadme.ch/index.php) was employed to predict ADME (Absorption, Distribution, Metabolism and Excretion) parameters, including bioavailability score, gastrointestinal (GIT) absorption and blood-brain barrier (BBB). Toxicity analysis was done by Toxtree (version 3.1.0.1851), a generic open source application, which is able to estimate toxic hazard by making predictions for a number of toxicological endpoints by different modules, such as carcinogenicity prediction (genotoxic and non-genotoxic) and Ames carcinogenicity tests.
Results
Screening of Shanmei Capsule active ingredients
A total of 59 candidate active ingredients were screened from the TCMSP, including 25 from Crataegi Folium, 29 from Dahurian rose fruit, and 5 shared ingredients. Details are shown in Table 1. The obtained candidate ingredients and all of the potential targets were applied to construct a network of ingredient-target interactions, including 330 nodes (59 ingredients and 271 targets) and 928 edges, as shown in Fig. 1. The degree of targets was adopted as a characteristic topological parameter to define the significance of candidate ingredients. The interactions indicated that one ingredient could regulate numerous targets, such as quercetin, kaempferol,
A list of the selected compounds in Shanmei Capsule for network analysis
A list of the selected compounds in Shanmei Capsule for network analysis
Establishment of component-target network. Components are shown as red dovetails nodes and target are marked as purple circular nodes.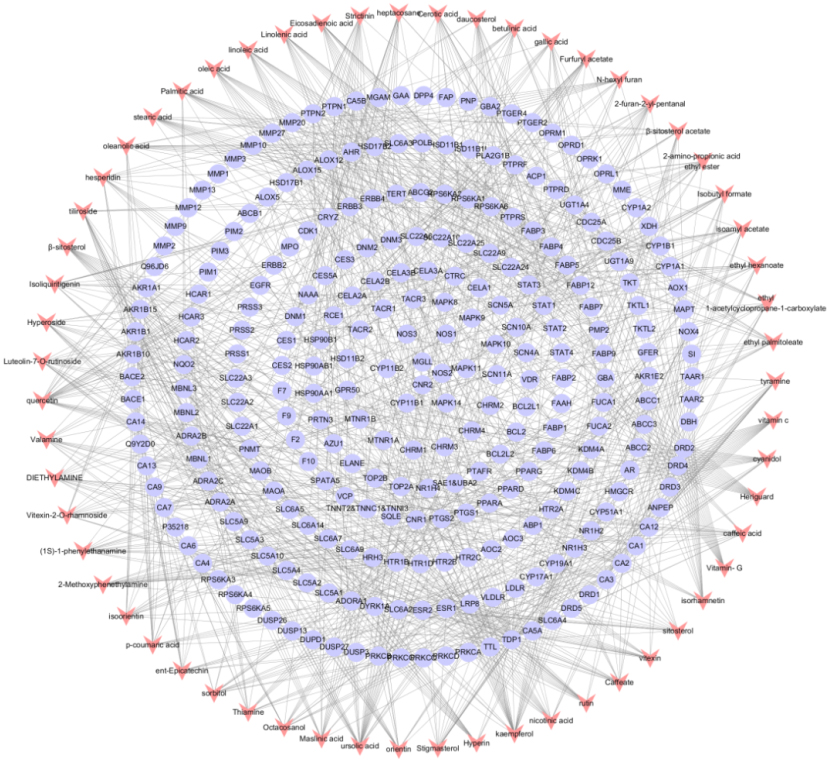
A total of 1018 potential targets to have correlations with hyperlipidemia were found via the GeneCards database. After merging hyperlipidemia-related targets and active ingredient targets, 34 overlapping targets were considered as candidate targets (Table 2, Fig. 2). Further linking of target ingredients with candidate targets based on Cytoscap 3.3.0 resulted in a network of disease-ingredient-target, with 51 nodes and 301 edges (Fig. 3). Among them, 17 red dovetail nodes represent active ingredients of Shanmei Capsule, and 34 purple ellipse nodes represent candidate targets of hyperlipidemia. After analyzing the topological characteristics of each node in the interaction network, it shows that the node degrees of eight targets, HMGCR, MMP9, MAPK8, PPAR
Related parameters of potential targets of Shanmei Capsule
Related parameters of potential targets of Shanmei Capsule
Target-target interaction network between active ingredients of Shanmei Capsule against hyperlipidemia.
Component-target-diseas interactive network of Shanmei Capsule. Red dovetails nodes are the main active ingredients of Shanmei Capsule, and purple circular nodes are the potential targets for treating hyperlipidemia.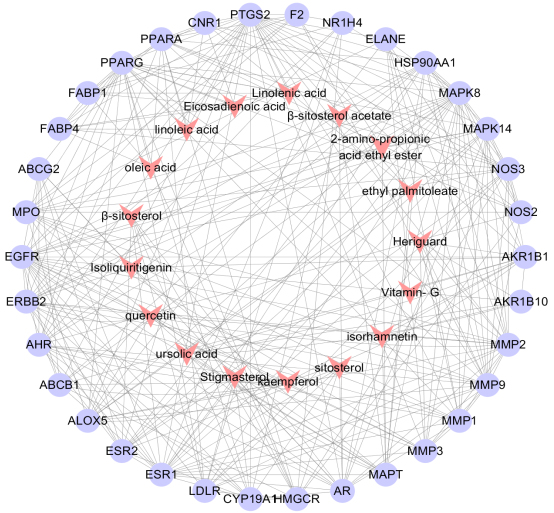
To further clarify relevant functions and pathways, GO functional analyses and KEGG pathway enrichment were performed for above 34 core targets using DAVID database. GO enrichment analysis was annotated in the following three categories, including biological process, cellular component and molecular function, with
Pathway enrichment of potential targets based on KEGG pathway analysis
Pathway enrichment of potential targets based on KEGG pathway analysis
Go and KEGG pathway enrichment bubble diagrams for candidate targets (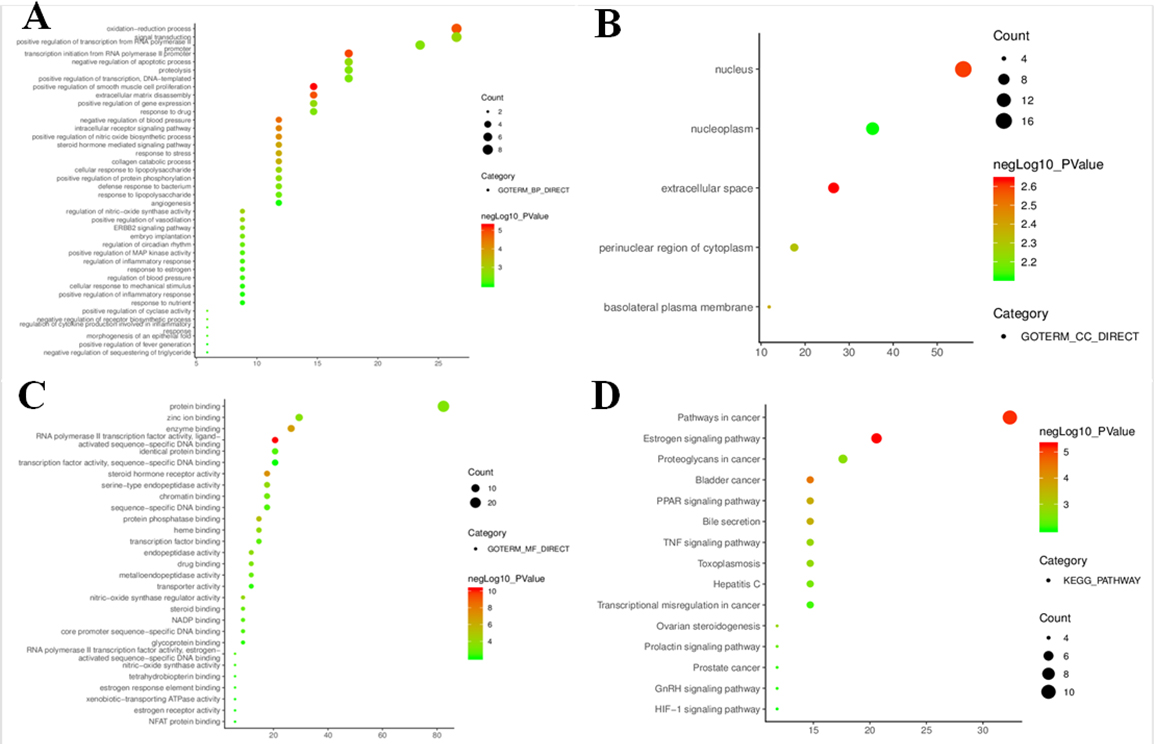
The component-target-pathway network for Shanmei Capsule. Purple circular nodes represent the candidate targets, red dovetails nodes represent the active ingredients and rose red parallelogram nodes represent the KEGG pathways. The size of nodes was proportional with the degree and larger sizes represent greater associations with hyperlipidemia treatment.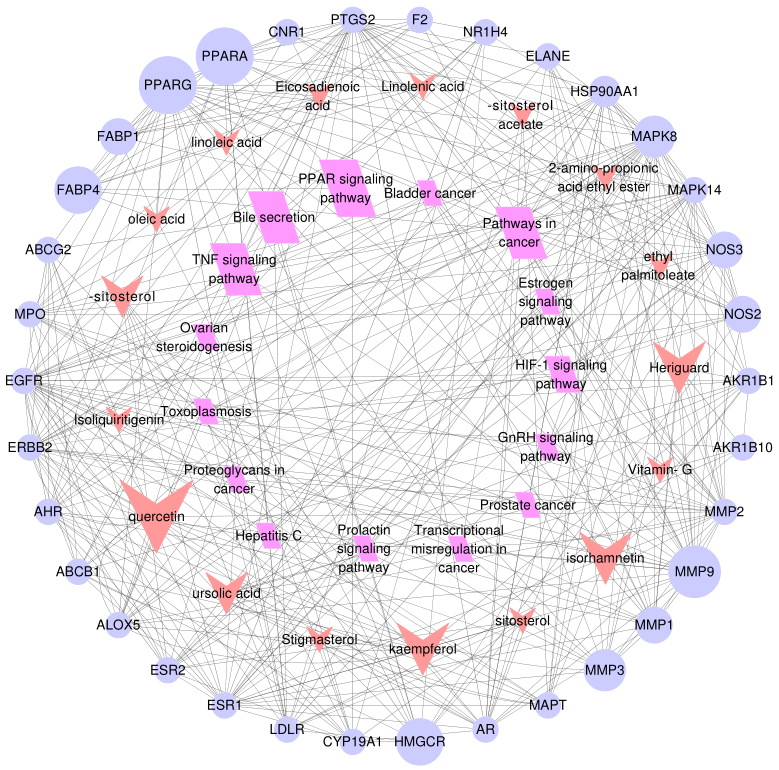
At last, in order to elucidate the connection between the core targets and corresponding pathways, a component-target-pathway interaction network map was generated by Cytoscape 3.3.0 (Fig. 5). The 17 red dovetails nodes represent the active pharmaceutical ingredient, the 34 purple circular nodes denote the potential targets, and the 15 rose red parallelogram nodes represent the pathways. And the node sizes depend on degree value. The larger the node is, the larger the degree value is. There were 66 nodes and 340 edges were involved in the association between these targets, which reflected the biological process of the key targets. These results were revealed that Shanmei Capsule take effect in hyperlipidemia treatment by multi-components, multi-targets and multi-pathways.
Molecular docking was performed among 17 ingredients and 34 potential targets using the Autodock Vina software, and detailed results are presented in Fig. 6. Affinity was the binding score for the molecular docking, and when the docking score value was smaller, the binding affinity to target protein was stronger. The docking score
The heat map of molecule docking scores.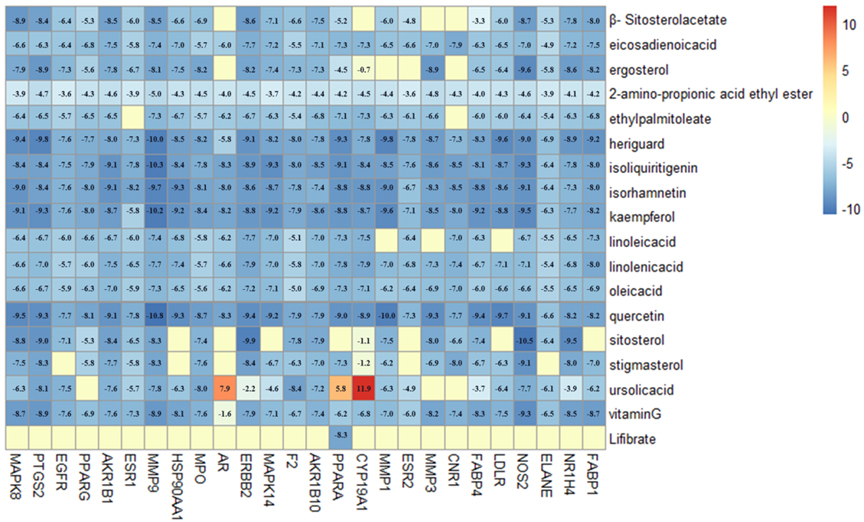
The ADME properties of 17 active ingredients were calculated by SwissADME web tool. As is can be observed in Table 4, all the ingredients, besides heriguard, have bioavailability score above the threshold of 0.5, which might suggest good oral bioavailability. The majority of the tested ingredients have no ability to penetrate the blood-brain barrier, thus presenting a low risk for central nervous system side effects. Based on toxicity tests, it appears that all the ingredients have no nongenetic carcinogen properties, with only vitaminG was predicted to be genetic carcinogen. The results of SwissADME calculations and toxicity tests provided useful information about the selected ingredients and found to be free from high risks of undesired effects.
ADME and toxicity prediction results of 17 key ingredients in Shanmei Capsule
ADME and toxicity prediction results of 17 key ingredients in Shanmei Capsule
Notes: (
Molecular models of bioactive ingredients and positive drug binding to the predicted PPAR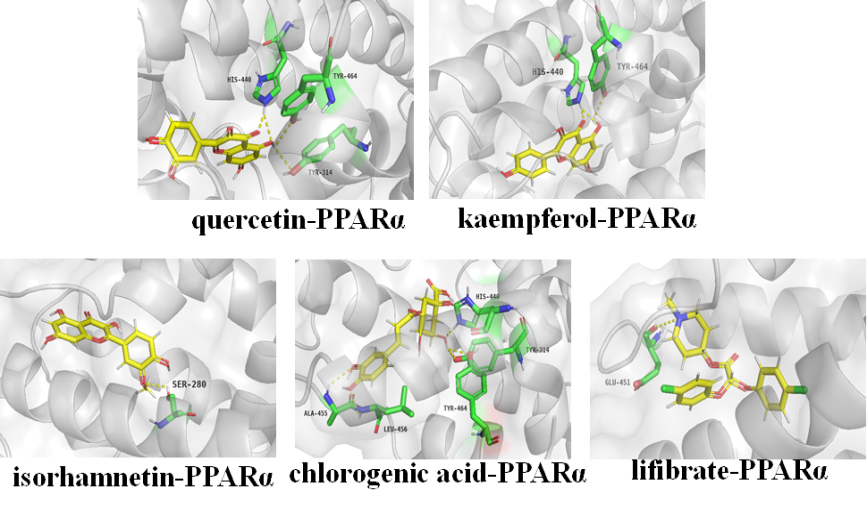
Hyperlipidemia is one of the most prevalent global chronic metabolic diseases, which usually involved in a disorder of lipid metabolism and accompanied by elevated contents of TC, TG, LDL-C, as well as decreased HDL-C levels. During this process, blood lipid level, oxidative stress, inflammation, and other factors might be the key link of the development of dyslipidemia-induced hyperlipidemia. Shanmei Capsule, a famous preparation in China, is made of a combination of Crataegi Folium and Dahurian rose fruit ethanol extract in proportion. Crataegi Folium and Dahurian rose fruit both are well-known traditional medicinal plants, which have been confirmed to possess various biological and pharmacological activities, such as anti-inflammatory, antioxidant and hypolipidaemic effects. Therefore, in this study, network pharmacology approach and molecular docking were applied to investigate the potential mechanism of multi-ingredient, multi-targets, and multi-pathway on Shanmei Capsule against hyperlipidemia (Fig. 8) [26, 27, 28, 29, 30].
Detailed signaling pathway of Shanmei Capsule in the treatment of hyperlipidemia.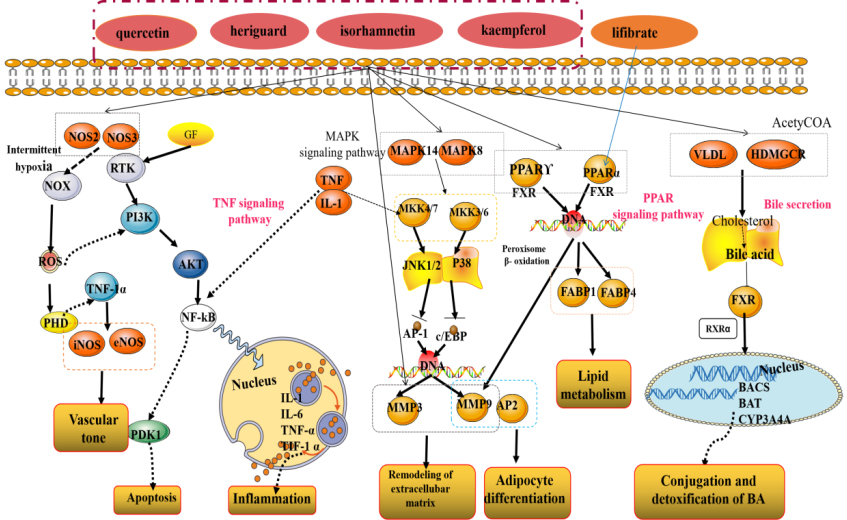
Via network pharmacology analysis, a total of 17 key active ingredients were regarded to be effective on hyperlipidemia, including quercetin, kaempferol, isorhamnetin, chlorogenic acid, ursolic acid and
Thirty-four potential targets consist of HMGCR, MMP9, MAPK, PPAR
The KEGG enrichment analysis indicated that the main pathways involved in the hyperlipidemia treatment process are PPAR signaling pathway, Bile secretion and TNF signaling pathway. PPAR signaling pathway participate in many physiological processes implicated in cardiovascular diseases associated with hyperlipidemia and diabetes, including lipid metabolisms, energy metabolisms, and inflammatory reactions, etc. PPAR
The potential binding sites and intermolecular interaction of active ingredients for target proteins were further elaborated by docking analysis. PPAR
In the present study, a network pharmacology approach in combination with molecular docking was proposed to explore the underlying mechanism of action of Shanmei Capsule against hyperlipidemia. The active ingredients of Shanmei Capsule in hyperlipidemia treatment are composed of 59 ingredients. Among them, quercetin, kaempferol, isorhamsin and chlorogenic acid are main active ingredients. The possible molecular mechanism mainly involved PPAR signaling pathway, Bile secretion and TNF signaling pathway via acting on MAPK8, PPAR
Footnotes
Acknowledgments
This work was supported by the Key Research and Development Program of Shanxi Province (201903D321205).
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Supplementary material
Grid parameters of the protein-ligand interaction for the key targets
Protein
PDB ID
Grid parameters of the protein-ligand interaction
Center x
Center y
Center z
Size x
Size y
Size z
AKR1B10
1ZUA
9
23.
9
15.
0
60.0
60.0
60.0
AR
2AX6
26.
9
3.
5
4.
4
17.8
18.5
12.5
CNR1
5TGZ
32.
3
19.
0
292.
2
39.8
73.1
70.4
CYP19A1
3EQM
85.
2
51.
3
42.
8
21.9
24.7
21.4
EGFR
5U8L
13.
4
7
9
21.7
29.1
47.6
ELANE
1H1B
10.
5
8.
9
15.
9
39.2
52.8
31.0
ERBB2
3PP0
16.
4
17.
4
26.
2
17.4
23.9
17.2
ESR1
3UUC
1
2
9
18.1
26.0
25.5
ESR2
3OLL
6
45.
0
10.
2
20.4
32.2
16.6
F2
3K65
22.
9
0.
9
2.
4
16.9
22.7
14.6
FABP4
5Y0F
5.
6
5
2
17.4
16.1
18.6
LDLR
1IJQ
1
39.
3
47.
6
50.0
48.0
52.0
MAPK8
4L7F
1
53.
8
4.
3
19.1
17.3
27.5
MAPK14
6SFI
53.
4
69.
4
17.
4
19.7
23.8
21.7
MMP1
966C
8.
8
9
38.
9
20.4
18.4
18.8
MMP3
1D7X
4
19.
8
29.
6
40.0
40.0
40.0
MMP9
4H1Q
29.
2
6.
0
18.
9
37.5
22.8
25.2
MPO
5MFA
5
4.
9
0.
3
64.4
65.2
84.7
NOS2
4NOS
3.
1
95.
2
17.
2
26.8
19.1
30.0
NR1H4
6HL1
10.
9
7
12.
3
17.9
23.7
14.5
PPARA
5HYK
8.
7
33.
9
19.
3
14.5
18.0
19.8
PPARG
6T9C
14.
8
17.
3
17.
7
21.9
17.8
18.8
PTGS2
5F19
28.
1
41.
0
56.
6
54.1
75.2
45.6
AKR1B1
5OUK
4.
5
2.
7
4.
8
27.0
24.7
20.5
FABP1
3STK
9
8
19.
4
24.2
18.4
28.0
HAP90AA1
4LWE
7
0
3
17.1
17.9
17.3