
Abstract
Parkinson’s disease (PD) is the second most common still relentlessly progressive neurodegenerative disorder with a long period in which the pathophysiological process is already spreading but cardinal motor symptoms are not present. This review outlines the major developments and milestones in our understanding of PD that have shaped the way we define this disorder. Past criteria and definitions of PD have been based on clinical motor manifestations enabling diagnosis of the disease only in later symptomatic stages. Nevertheless, with advancing knowledge of disease pathophysiology and aim of early disease detection, a major shift of the diagnostic paradigm is being advocated towards a biological definition similar to other neurodegenerative disorders including Alzheimer’s disease and Huntington’s disease, with the ultimate goal of an earlier, disease course modifying therapy. We summarize the major pillars of this possible approach including in vivo detection of neuronal α-synuclein aggregation, neurodegeneration and genetics and outline their possible application in different contexts of use in the frame of biological PD definition.
FROM THE PHENOTYPE TO UNDERLYING BIOLOGY: EVOLUTION OF CONCEPTS
Parkinson’s disease (PD) is the second most common neurodegenerative disorder [1], yet the journey of discovering the underlying pathomechanisms leading to the classic motor phenotype was a long and complicated one, with many questions still unsettled. Our understanding of PD pathophysiology evolved enormously during the past 200 years and the changes in diagnostic criteria reflect this development. The most significant shift in defining PD is the currently proposed transition from a clinical towards a biological definition of the disease. To map this process, it is important to start with a historical excursion outlining the most important research milestones that shaped our understanding of what is (and is not) PD.
On the track of PD-related neurodegeneration: from the “shaking palsy” to α-synuclein and beyond
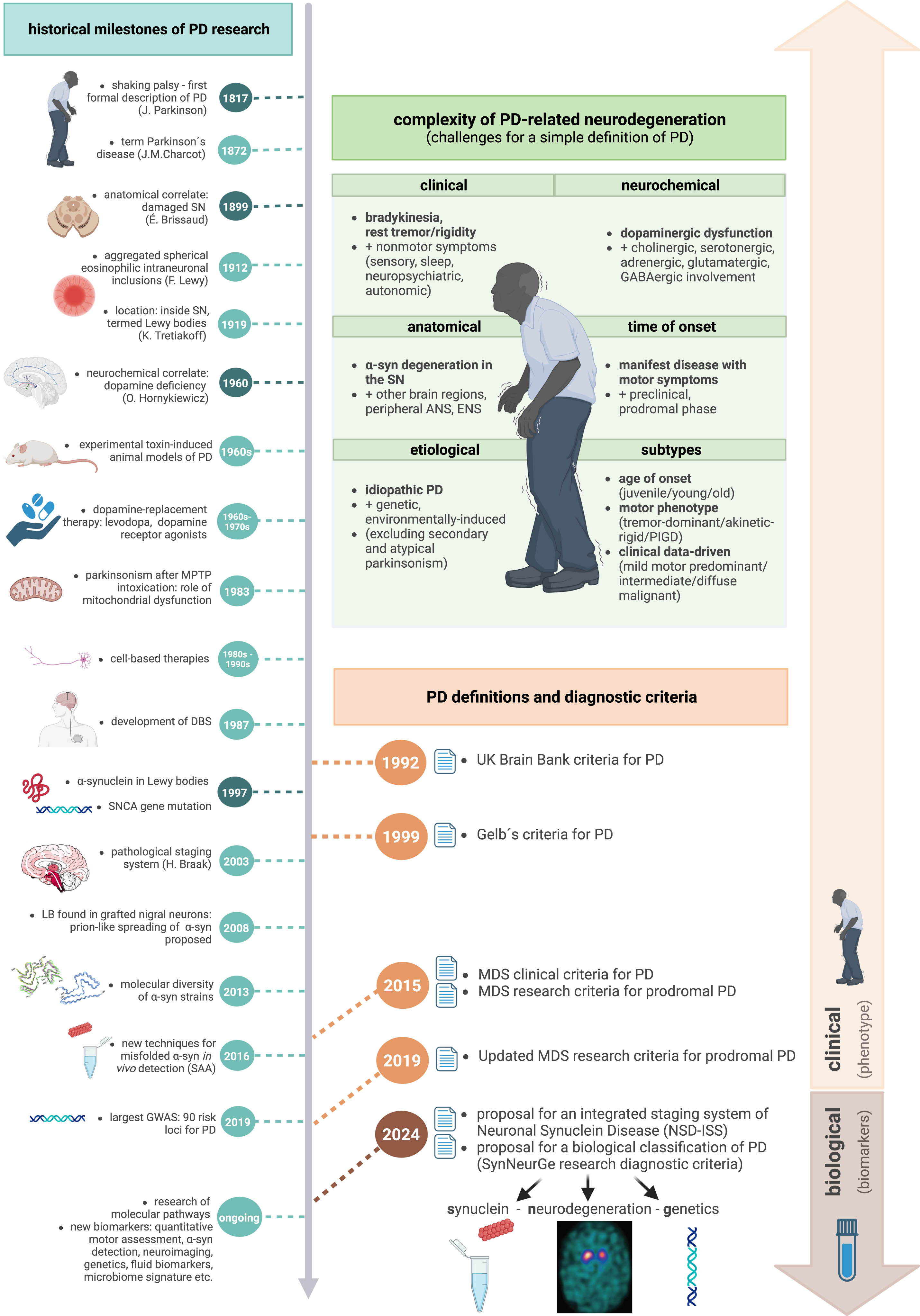
Although there have already been several records reporting PD-like symptoms throughout the centuries [2, 3], it was only in 1817 when the disease was formally described by James Parkinson in “An Essay on a Shaking Palsy” [4], focusing on the motor manifestation – rest tremor, slowness or absence of voluntary movements, stooped posture and festinating gait. The term ‘Parkinson’s disease’ was proposed in 1872 by Jean-Martin Charcot instead of shaking palsy, acknowledging that these patients are not weak and not all of them present with tremor, along with the recognition of two basic subtypes, the tremulous and the rigid/akinetic form [5]. An anatomical correlate of damaged substantia nigra (SN) was discovered by Édouard Brissaud only in 1899 [6]. From histological perspective, Frederick Lewy described aggregated spherical eosinophilic intraneuronal inclusions in 1912 [7], followed by Konstantin Tretiakoff locating these structures inside the SN and naming them Lewy bodies (LB) in 1919 [8]. 40 years later, a neurochemical correlate of dopamine deficiency in the striatum of PD patients was found by Oleh Hornykiewicz in 1960 [9], soon leading to the introduction of a dopamine-replacement therapy: orally administered levodopa and first dopamine receptor agonists [10–12]. This treatment is still so effective that sustained response to levodopa remains a supportive criterion for PD even in the current diagnostic criteria. After a period, in which neurosurgical pallidotomy was applied to relieve severe symptoms in late stage PD [13], a better understanding of the electrophysiological correlates of basal ganglia circuits dysfunction and the excessive synchronized oscillation [14] ultimately led to development of deep brain stimulation (DBS) of several targets (the ventral intermediate nucleus of the thalamus in 1987 [15], followed by the globus pallidus internus and subthalamic nucleus [16]), alleviating motor symptoms of PD. In the meantime, several experimental toxin-induced animal models of PD were established in the 1960s, replicating nigrostriatal dopaminergic dysfunction and enabling further study of PD pathogenesis, e.g., the 6-OH-dopamine model [17]. In early 1980s, serendipitous observation of acute onset of parkinsonism following illicit drug abuse (1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine – MPTP, due to inhibition of complex I of the electron transport chain) brought the role of mitochondrial dysfunction into research spotlight [18]. Multiple other cellular and molecular pathways have been recognized since then, including impaired protein degradation mechanisms, oxidative stress and neuroinflammation [19]. Several cell-based therapies were conducted in PD patients between the 1980s and 1990s, based on data from preclinical models showing fetal mesencephalic tissue-derived cell suspensions reinnervating the host striatum and restoring dopaminergic deficits in animal models [20]. A major breakthrough in the understanding of the molecular origins of PD came with the discovery of α-synuclein (α-syn) as the most important component of Lewy bodies in 1997 [21], together with the identification of a SNCA gene mutation as the first gene causing monogenic PD in the same year [22]. Thus, the α-syn era of PD emerged. After the reports in 2008, stating that Lewy bodies were found in grafted nigral neurons 14 years post transplantation in several PD individuals [23], a prion-like spreading of misfolded α-syn was proposed, supported by findings from experimental animal models [24]. Currently, a molecular diversity of α-syn strains is being recognized, suggesting an explanation for the clinical heterogeneity of various α-synucleinopathies beyond PD [25]. Development and validation of different techniques for misfolded α-syn in vivo detection is ongoing [26].
Further genetic discoveries of new monogenic forms of PD over the past years unraveled several more mechanisms and common biological pathways contributing to PD pathogenesis. The most important impaired biological processes include mitochondrial quality control and regulation (linked mostly to recessive mutations in PINK1, PRKN, DJ-1, but also FBXO7, POLG, VPS13C, CHCHD2, and LRRK2 genes), autophagy–lysosomal pathways (ATP13A2, GBA1, LRRK2), endocytic membrane trafficking pathways (VPS35), cell membrane (PLA2G6), immune response (both inflammation and autoimmune response; associated with BST1 and HLA genes), synaptic vesicle formation and trafficking (DNAJC6, SYNJ1) or microtubule function (DCTN1, LRRK2) [27, 28]. These findings are particularly important for drug development in the upcoming era of precision medicine, directly targeting affected pathways. On the other hand, sporadic PD is caused by a complex interplay of genetic and environmental factors (the “exposome”), including epigenetics, which is increasingly considered to have a potential role in PD pathogenesis [29]. Gene–environment interactions may involve synergistic effects between genetic variants and specific risk factors of PD (such as diabetes, smoking, caffeine intake, head trauma or pesticide exposure) [30, 31]. Between the poles of monogenic PD vs. common variants, there are uncommon gene variants with intermediate effect, which have variable penetrance (LRRK2/GBA-PD). In case of LRRK2, which may contribute to PD through mitochondrial function, autophagy and/or microtubule stability, the G2019S variant has an estimated age-dependent PD penetrance of 25–74% [32]. This incomplete penetrance may be related to both environmental (e.g., smoking, non-steroidal anti-inflammatory drugs) and genetic modifiers [28]. Heterozygous GBA variants encoding lysosomal enzyme β-glucocerebrosidase are linked with a PD penetrance ranging between 10–30%. Different severity of variants and other genetic modifiers (such as LRRK2, TMEM175, SNCA, and CTSB) may lead to variable phenotypic severity [33]. Rapid advances in the field of PD genetics continue to untangle hidden pathogenetic pathways. However, genome-wide association studies (GWAS) and other genetic techniques have not yet been able to explain most of the observed heritability (as seen in the largest PD GWAS where 90 risk loci were identified, explaining only 16–36% of the heritable risk of PD) [34]. This missing heritability, common to many complex diseases, may continue to be reduced as the role of non-coding genetic variants is anticipated [35].
An overview of the most important historical advances in understanding PD pathophysiology is displayed in Fig. 1.
Widening the picture: complexity of Parkinson’s disease, challenges, and controversies
The narrow and straightforward definition of PD has so far been centered around the typical motor phenotype of bradykinesia plus one or both of rigidity and/or resting tremor. These PD defining motor symptoms are caused by progressive nigrostriatal dopaminergic degeneration driven by underlying α-synucleinopathy, with histological findings of Lewy bodies and accompanying pathophysiological processes. While these PD hallmarks remain true for most cases, the story of PD became more complex over decades of research, posing significant challenges for a simple definition.
From clinical viewpoint, multiple non-motor symptoms expand the phenotype of PD: sensory (e.g., hyposmia, pain, visual and somatosensory disturbances), neuropsychiatric (e.g., anxiety, depression, apathy, fatigue, dementia, psychosis), sleep (e.g., REM sleep behavior disorder – RBD), and autonomic (e.g., urinary, erectile dysfunction, orthostatic hypotension, constipation) [36]. From the neurochemical perspective, other neurotransmitter systems beyond dopaminergic are involved, such as cholinergic, serotonergic, adrenergic, glutamatergic, or GABAergic [37]. As for the anatomy, α-syn-based degeneration affects many regions of the nervous system apart from the substantia nigra – not only other brain regions, but also the peripheral autonomic and enteric nervous systems (ANS, ENS), salivary glands and skin [38]. For the differential diagnosis of PD versus other causes of parkinsonism, secondary forms (e.g., vascular, infectious, drug/toxin-induced, metabolic, posttraumatic or related to normopressure hydrocephalus), atypical forms (progressive supranuclear palsy – PSP, corticobasal degeneration – CBD, multiple system atrophy – MSA) and several hereditary neurodegenerative disorders with parkinsonism as one of possible manifestations (e.g., Wilson’s disease, Huntington’s disease, etc.) need to be considered [39]. Excluding these causes, we end up with an umbrella term of idiopathic PD (iPD) for still a significantly heterogeneous group of patients, including opposite sides of the “nature vs. nurture” spectrum—cases with a likely environmental contribution to the pathogenesis (e.g., by pesticides) [40], as well as cases of monogenic PD (accounting for up to 5–10% of all PD cases) [41]. Moreover, several PD-linked gene variants such as PRKN, PINK1, or LRRK2 exhibit almost no or only limited α-syn pathology [42]. It also became evident that co-pathologies can influence disease manifestation. In this respect dementia with Lewy bodies (DLB) is seen as part of the PD spectrum, in general associated with prominent cortical spreading of the α-syn pathology and additional vascular and/or Aβ pathology [43, 44].
A broad heterogeneity of clinical manifestations even within the current iPD group elicited an effort to further classify PD phenotypes. Several attempts have been made—based on age of onset (juvenile/young/old), motor phenotype (tremor-dominant/akinetic-rigid/postural instability and gait disturbance – PIDG), clinical data-driven approach (mild motor predominant/intermediate/diffuse malignant), or the first occurrence of α-syn misfolding and further temporal sequence of symptoms onset (body-first/brain-first) [42, 46], with a partial overlap between them (e.g., PIGD subtype with more severe progression, higher load of non-motor symptoms, higher prevalence of dementia and hallucinations is consistent with diffuse malignant and body-first subtype). However, validity and widespread applicability of these data-driven PD subtype classifications are still questioned due to limited reproducibility [47].
This complexity and heterogeneity makes it challenging to create a uniform definition applicable for all abovementioned cases. Controversies are the consequence as illustrated by the following three exemplary cases: 1) a case of Parkin-PD with young onset, benign phenotype, slow progression, minimal cognitive impairment, limited or no α-synucleinopathy, 2) a case of PD with diffuse malignant subtype with fast progression, presence of RBD, α-syn misfolding, and dementia onset 1.5 year after the diagnosis, and 3) a case of DLB with fast progression, presence of RBD, α-syn misfolding, and dementia onset within 1 year. The first two are currently termed idiopathic PD, although their clinical manifestations and underlying biology are significantly different, while the last one is still considered by many as different from the first two despite almost identical clinical and biological profiles with the second one.
Another issue is the time point of diagnosis. Neurodegeneration in PD is a slow and gradual process starting years or even decades before evident parkinsonism is present. Pioneer work of Heiko Braak described α-syn accumulation first occurring in lower brainstem (stage 1), with later affection of the SN (only in stage 3), ultimately leading to cortical deposition (stage 6), described as Braak stages and explained by progressive ascendent spreading of α-syn from the periphery (likely ENS via vagus nerve) [48]. Although other trajectories were proposed later [49, 50], their shared observation is the beginning of α-syn misfolding outside of the SN, often outside the CNS, directing research attention to these phases of early neurodegeneration, with the perspective of future disease-modifying trials. Currently, neurodegeneration in PD is classified into three phases: 1) the preclinical phase, which represents a period of ongoing neurodegeneration without any detectable symptoms (although with biomarkers positivity), 2) the subsequent prodromal phase, characterized by a combination of mostly non-motor and in its later part also early motor symptoms, and 3) the clinical phase, defined by the characteristic motor symptoms [51]. RBD is the strongest prodromal predictor of PD, with a more than 80% risk of converting into an overt α-synucleinopathy in the following years [52]. In this scenario, PD is perceived as a continuum and posing strict boundaries between prodromal and manifest PD seems artificial. Thus, the question arises: which features of the neurodegenerative process are necessary (or sufficient) for the definition of the disease? In other words, is RBD already PD? Given the underlying biology, the answer is likely yes (in most cases), however, considering clinical diagnostic criteria, not yet.
DEFINING PD: PAST AND CURRENT CRITERIA
After the first description of PD, no formal diagnostic criteria were available for more than 150 years until the late 1980s. A clinicopathological definition, widely agreed on by both scientists and clinicians, described PD as a slowly progressive neurological disorder with parkinsonism without features suggestive for an alternative diagnosis, responding to dopaminergic treatment, and associated with loss of SN neurons and the presence of LBs in some of the remaining neurons [53].
Overview of past, current, and proposed diagnostic criteria for Parkinson’s disease
SN, substantia nigra; UK, United Kingdom; PD, Parkinson’s disease; y., year; CT, computed tomography; MPTP, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine; LD, levodopa; dg., diagnosis; LB, Lewy bodies; MDS, International Parkinson and Movement Disorders Society; MIBG, metaiodbenzylguanidin; bvFTD, behavioral variant of frontotemporal dementia; PPA, primary progressive aphasia; pPD, prodromal Parkinson’s disease; LR, likelihood ratio; RBD, REM Sleep Behavior Disorder; PET, positron emission tomography; SPECT, single-photon emission computerized tomography; GF+, fully penetrant pathogenic gene variants; GP+, pathogenic gene variants with strong or intermediate predisposition; G–, gene variants – indeterminate; α-syn, alpha-synuclein; SAA, seed amplification assays; CSF, cerebrospinal fluid; ICH, immunohistochemistry; IF: immunofluorescence; FDG, fluoro-deoxy-glucose; MRI, magnetic resonance imaging; MSA, multiple system atrophy; PSP, progressive supranuclear palsy; S+/S–, presence/absence of synucleinopathy; N+/N–, presence/absence of neurodegeneration; Cposs+, clinical manifestations possibly related to PD; Cprob+, clinical manifestations probably related to PD; PSG, polysomnography; SynNeurGe, synuclein-neurodegeneration-genetics; NSD-ISS, integrated staging system of neuronal alpha-synuclein disease; D+/–, presence/absence of dopaminergic dysfunction/degeneration.
Starting with the UK Parkinson’s Disease Society Brain Bank Diagnostic Criteria (UK Brain Bank Criteria) in 1992, several sets of clinical diagnostic criteria were used during the last decades (for overview, see Table 1). The UK Brain Bank Criteria were based on a clinicopathological correlation of histopathological findings from 100 iPD patients and consisted of the diagnosis of parkinsonism (defined as bradykinesia + at least one of rigidity/rest tremor/postural instability), absence of exclusion criteria, and presence of at least 3 supportive criteria [54]. Gelb’s diagnostic criteria for PD (1999), based on a literature review, proposed three levels of diagnostic confidence: possible PD (≥2/4 core motor features of parkinsonism with obligatory bradykinesia/tremor, absence of features suggestive of alternate diagnosis/duration < 3 years, response to dopaminergic treatment/patient has not had an adequate trial of levodopa or dopamine agonist), probable (≥3/4 core motor features of parkinsonism, absence of features suggestive of alternate diagnosis, response to dopaminergic treatment) and definite (all criteria for possible PD met, together with histopathologic confirmation, defined as depletion of neurons in the SN, presence of LB and absence of alternate diagnosis) [55].
In 2015, the MDS clinical diagnostic criteria for PD were introduced, designed for research purposes, but applicable also in clinical practice [43]. After determining the presence of parkinsonism (defined as bradykinesia + rigidity/rest tremor), two levels of diagnostic certainty were proposed – clinically established PD (based on absence of absolute exclusion criteria, presence of ≥2 supportive criteria, and no red flags) and probable PD (based on absence of absolute exclusion criteria and presence of red flags counterbalanced by supportive criteria –1 supportive criterion is required in case of 1 red flag, ≥2 supportive criteria are needed in case of 2 red flags, >2 red flags are not allowed). Compared to the UK Brain Bank criteria, the MDS-PD criteria performed better in differentiating cases of atypical or secondary parkinsonism from iPD [56].
To enable detection of PD patients before the onset of evident parkinsonism, MDS research criteria for prodromal Parkinson’s disease (pPD) were proposed in the same year [57]. These criteria assess presence or absence of several risk factors and prodromal markers including male sex, pesticide and solvent exposure, non-use of caffeine, non-smoking, family history of PD/known gene mutation, SN hyperechogenicity, RBD, abnormal dopaminergic imaging (PET/SPECT), possible subthreshold parkinsonism, hyposmia, constipation, excessive daytime somnolence, symptomatic hypotension, erectile dysfunction, urinary dysfunction, and depression (±anxiety). In case of presence/absence of given markers, a positive/negative likelihood ratio (LR) is attributed to each of them and by multiplying all LRs, a total LR is obtained, which is then combined with baseline pretest age-dependent probability to calculate final post-test probability of pPD applying a Bayesian statistic formular. In case of exceeding the threshold of ≥80% probability, probable pPD is diagnosed. Meeting the lower threshold of ≥50% probability falls into the category of possible pPD. An update of the MDS pPD research criteria was published in 2019 [58], adjusting numeric values of LRs of already included risk/prodromal markers, introducing refined subcategories (genetic background, neurogenic orthostatic hypotension) and adding several new markers (low plasmatic urate level in men, physical inactivity, diabetes mellitus type 2, global cognitive deficit).
Although the MDS criteria have been widely accepted and used during past years, there are still several challenges. As for MDS-PD criteria, they are clinically oriented and do not reflect sufficiently underlying biology (no histopathological confirmation, absence of molecular markers). MDS pPD criteria pay more attention to non-motor symptoms and show overall good specificity in validation studies when applied in enriched cohorts (e.g., [59–61]), although they struggle with low sensitivity in the general population if specific markers with high LR (DaTscan, polysomnography-confirmed RBD) are not included (e.g., [60, 62]). As they are heavily weighted towards RBD-positive prodromal subtype, their applicability for other pPD subtypes with less extensive load of non-motor symptoms is rather limited [63].
Regarding a clinical staging system of PD, the Hoehn and Yahr (H&Y) scale was developed in 1967 and continues to be broadly applied until now, anchored at the distinction between unilateral disease (stage I) and bilateral disease (stages II–V) and the development of postural reflex impairment (stage III) as a key turning point in the disease’s clinical significance [64]. Despite its broad use in describing the overall motor burden and the progression of functional disability, it rates based upon only a small fraction of important PD symptoms (driven predominantly by symmetry and gait) [65]. Moreover, its use is limited to the late stages of PD with clinically manifest parkinsonism, making it a tool for assessing impact of motor impairment, not staging of PD-related neurodegeneration as such.
FROM CLINICAL TO A BIOLOGICAL DEFINITION OF PD
With a growing body of knowledge on the pathophysiological background of PD and advances in biomarker research, especially techniques for in vivo detection of neuronal α-syn (n-α-syn) aggregation, a shift of the diagnostic paradigm from a clinical to a biological one is currently being advocated. Clinical PD/pPD diagnostic criteria are dependent on the presence of clinical symptoms and thus are applicable only in later/more advanced symptomatic stages of the disease. Prodromal criteria help to detect earlier stages, but again rely upon clinical symptoms and signs. On the other hand, biological diagnostic criteria could potentially cover the earliest presymptomatic stages of the disease and enable clustering of biologically homogeneous patients. Biological definitions could have major consequences for future research and clinical advancements, especially towards early therapeutic interventions and precision medicine. Biological definition of PD is analogous to recent developments in other neurodegenerative disorders. These include Alzheimer’s disease with the amyloid, tau, neurodegeneration (A/T/N) classification system. This was based on proteinopathy-based imaging, cerebrospinal fluid (CSF) biomarkers, and structural imaging, upon which clinical features are added once the biological definition is made [66, 67]. The Huntington’s disease Integrated Staging System (HD-ISS) similarly defines a biological research definition and staging centered on genetic, neuroimaging, biological, clinical, and functional assessments [68].
Two proposals for the biological classification and/or staging of PD have been recently made [69–71]. Both center around α-synucleinopathy and evidence of neurodegeneration, but with important differences. One system (the SynNeurGe research diagnostic criteria, acknowledging the molecular complexity and heterogeneity of PD) proposes a biological S/N/G classification, which proposes that PD can be classified with documentation of α-synucleinopathy (either from immunohistochemistry (IHC) or α-syn seed amplification assays (α-syn SAA)), presence of associated neurodegeneration (either peripheral or central), and genetics (e.g., documentation of PD-related gene can allow classification of PD even in the absence of α-synucleinopathy). Once biological diagnosis has been made, clinical symptoms can be assessed, and then attributed as either probably or possibly due to PD, depending upon diagnostic strength [69]. The second system (intended to be a research framework to enable interventional trials at early disease stages, targeting α-syn), rather than staging PD per se, defines “Neuronal α-Synuclein Disease” (NSD) encompassing both PD and DLB. This has two primary criteria, namely documentation of α-syn (in this case only with cerebrospinal α-syn SAA), and dopamine denervation (via dopaminergic functional imaging). Staging further is done by rating functional impairment, resulting in an integrated staging system of NSD (NDS-ISS) [70]. While the approaches agree on the centrality of α-syn and neurodegeneration, differences in criteria can have important implications for diagnosis and clinical trials in PD. These approaches are currently discussed and will certainly be further developed with advancement in biomarker research. In the following section, a brief description of the primary components of biological classification systems is summarized.
Biological classification: α-synucleinopathy
Arguably the biggest step towards a biological definition of PD was advancement in techniques to detect pathological n-α-syn accumulation in vivo, especially α-syn SAA [72]. CSF α-syn SAA has been validated as a robust biomarker of n-α-syn with >90% of autopsy-confirmed PD and DLB cases being positive [73–75]. It also detects n-α-syn in iRBD, pure autonomic failure, hyposmia and non-manifesting carriers of monogenic PD [75]. In addition to CSF, SAA in skin biopsies shows a diagnostic accuracy of >90% in differentiating PD and iRBD versus healthy controls [76]. Other promising fluids/tissues including neuron-derived exosomes in blood [77] and others [76] are being evaluated. Importantly, SAA seems able to distinguish n-α-syn in Lewy body disease from other forms of α-syn, such as the predominantly glial pathology in MSA [78]. In addition to SAA, IHC detection of phosphorylated n-α-syn in skin biopsies shows high specificity to distinguish PD and iRBD from healthy controls, although with somewhat lower sensitivity. Skin IHC also differentiates PD from MSA [79, 80]. The diagnostic yield of α-syn IHC in other tissues, such as submandibular gland or colon is more variable [80–82].
Interpretation of S+ status in classification systems can vary. In case of NSD-ISS criteria, S+ status is a necessary component, except for carriers of fully penetrant gene variants unequivocally predisposing for α-synucleinopathy such as SNCA. Conversely, in the SynNeurGe classification, S–status does not preclude PD, particularly in cases of monogenic PD related to selected pathogenic variants such as LRRK2, PRKN, or PINK1. Nonetheless, both systems are challenged by false negative S+ assays, and by potential new PD genes remaining to be discovered, which will not be included in the biological PD classification. Furthermore, implications of an isolated S+ test warrant careful consideration as the positivity rate of SAA in asymptomatic older adults has been estimated at around 5–10% [75]. It remains unclear whether all those with a S+ test have true Lewy body disease, as opposed to possible transitory S+ states that are successfully cleared from the CNS. While NSD-ISS criteria recognize asymptomatic S+ individuals as already having defined, early stage 1A of the disease (although stressing the need for longitudinal studies assessing the incidence of and rate of progression to D+ and functional impairment) [70]; SynNeurGe criteria maintain a more cautious approach by classifying these individuals as having Parkinson’s type synucleinopathy (not yet sporadic PD), thus acknowledging the uncertainty of eventual developing clinical disease [69].
Biological classification: neurodegeneration
Neurodegeneration is a fundamental biological feature in all PD cases. Confirmation of PD-associated dopamine neurodegeneration can be obtained using dopamine transporter (DaT), vesicular monoamine transporter (VMAT2), or aromatic acid decarboxylase (F-dopa) SPECT/PET [83]. Studies consistently demonstrate that DaT loss typically occurs prior to functional motor impairment, at least in classic PD. Subjects with prodromal PD and abnormal DaT SPECT are likely to develop functional motor impairment within 3 to 5 years [84]. Although these abnormalities are highly sensitive for classic PD changes, they lack specificity as striatal dopaminergic denervation also presents in other neurodegenerative parkinsonisms, including MSA, PSP, and CBD [85] and even other neurodegenerative disorders like Huntington’s disease [86] or Wilson’s disease [87].
Other imaging markers of neurodegeneration can be documented in PD concurrently or even prior to striatal denervation [88], including specific patterns of brain glucose metabolism on FDG PET [89], cardiac sympathetic denervation on MIBG SPECT [88, 90], and specific MRI sequences sensitive to neuromelanin, iron levels, free water in the SN, and diffusion tensor imaging [85]. These markers vary in their sensitivity and specificity for differentiating PD from other parkinsonian syndromes and from healthy controls. Their application within the biological criteria is likely context-dependent, particularly in relation to disease stage. It is likely that S+ will usually appear prior to detectable neurodegeneration (hypothesis supported by limited but accumulating data [75], although more evidence is required), and the positivity of imaging methods may vary between different temporal patterns of neurodegeneration and/or biological subtypes of PD. So, according to the proposed criteria, those with detectable n-α-syn can potentially be diagnosed with NSD (stage 1A in the NSD-ISS criteria) or Parkinson’s type synucleinopathy (SynNeurGe criteria) even without neurodegeneration signs, although longitudinal studies are needed to assess future conversion rates of these S+ asymptomatic individuals, as discussed in the previous section. Conversely, without detectable n-α-syn, documentation of neurodegeneration would lead to PD diagnosis only in combination with presence of PD-related pathogenic variants such as LRRK2, PRKN, or PINK1, based on SynNeurGe criteria [69].
Biological classification: genetics
Genetic architecture of PD is complex, ranging from common variants individually contributing only a small amount to the PD risk [34], through uncommon, but not rare variants exerting an intermediate risk (e.g., GBA and LRRK2 variants), to rare, highly penetrant pathogenic variants leading to monogenic PD on the opposite side of the spectrum [27, 28], as outlined in more detail in the historical overview mapping genetic discoveries. Reported prevalence of monogenic PD differs greatly depending on the context of the studies. This has been ranging between 10–15% in the pre-selected PD populations from tertiary referral centers (e.g., ROPAD study [91]) and 1% in population-based studies with a representative case-mix [92], which seems more accurate and representative for general PD populations.
In the biological classification, with fully penetrant variants (SNCA monoallelic triplication, SNCA monoallelic pathogenic variants, PRKN, PINK1, and PARK7 biallelic pathogenic variants [69, 93]), PD may be diagnosed even without evidence of n-α-syn or neurodegeneration, similar to the recently proposed concept of Huntington’s Disease-ISS of Stage 0 [68]. In case of variants with incomplete penetrance (pathogenic variants in LRRK2, GBA1, VPS35, CHCHD2, or SNCA duplications [93]), additional proof of pathological n-α-syn accumulation or neurodegeneration would be required. In the SynNeurGe biological classification, fully penetrant S- genes are classified as PD if neurodegeneration is present. For the NSD-ISS criteria (which have α-synucleinopathy as their core), S- cases (LRRK2, PRKN, PINK1, etc.) are not classified [70, 93].
Clinical manifestations in the future era of biological PD definition
Even in the era of dominantly biological diagnostic criteria, clinical manifestations will still play a key role in sub-classification and monitoring progression of PD. Currently the dichotomy between prodromal and manifest PD (with “phenoconversion” defining the transition), is difficult to define in this continuous disease process. Future clinical stages might include an asymptomatic stage, followed by a clinical phase with symptoms possibly or probably associated with the disease, then stratifying clinical progression based both upon functional impact and/or new occurrence of hallmark clinical symptoms [69]. A version of this is reflected in the proposed NSD-ISS [70], in which stage 0 corresponds to a fully penetrant SNCA variant without any markers of neurodegeneration (similar to stage 0 in HD-ISS [68]), followed by stage 1 and 2 defined by positivity of misfolded α-syn detection and/or documentation of dopaminergic denervation. Clinical signs or symptoms can occur in stage 2, but stage 3 defines the state at which functional impairment occurs, which is further staged based upon severity until stage 6. Staging beyond stage 2 can only proceed if dopaminergic denervation has been documented (i.e., any functional impairment among DLB, RBD, or MCI patients without DaT abnormalities does not contribute to ratings beyond stage 2). As of the beginning of 2024, there is an ongoing scientific discussion in the scientific and clinical communities whether a biological classification or a staging system is indeed the best way to proceed. It is hoped that a consensus solution will be found, possibly integrating the most helpful and future-oriented aspects of both approaches. Moreover, it must always be kept in mind that any new definition and staging approach will require constant re-thinking and amendments as knowledge advances [94].
Finally, it is essential to consider that accessibility of several key components of potential biological criteria for PD (genetic testing, α-syn SAA, more sophisticated imaging methods) is low in many global health care settings. Therefore, clinical diagnostic criteria will continue to be widely used in routine practice for the foreseeable future.
FUTURE PERSPECTIVES AND UNMET NEEDS
A shift to a biological classification or definition of PD offers an unprecedented opportunity to advance our understanding of the disease. In contrast to the current concept, it potentially offers a definition of biologically more homogeneous patient subgroups, sharing similar pathophysiological background and thus enabling a more personalized management approach. While the current status of biological markers offers a rather categorical evaluation of individual domains (present/absent), there is a major need for identification of quantitative biomarkers to enable a better understanding and assessment of disease progression and response to therapy. These markers may also add to a staging approach, which will then more objectively discern different stages of the disease. This is crucial for disease-modifying or neuroprotective trials even in clinically asymptomatic stages of the disease with a higher likelihood of intervention effectiveness. In addition, as PD presents pathophysiologically a rather heterogeneous disorder, biomarkers that reflect underlying mechanisms of the disease (i.e., mitochondrial, inflammatory, lysosomal, and others) need to be developed to enable deeper biological subtyping and targeted management. These biological markers, along with some other promising candidates, such as microbiome signatures, other -omics analyses, wearables, and sensor-based markers, are likely to be incorporated in the future diagnostic criteria in case of sufficient supporting evidence. Lastly, to be generally applicable and globally available, further development needs to be directed towards biomarker platforms that will be broadly feasible, minimally invasive and cost-effective.
Presently, both sets of biological classifications (SynNeurGe, NSD-ISS) are designed to be used for research purposes. Future application of biological classifications will depend not only on the accumulating evidence on their reliability and diagnostic accuracy, but also on the technical advances, standardization, and availability of the biological tests themselves. Until then, the current clinical criteria for PD and pPD remain unchanged and generally recommended. Particularly in low-resource settings, where many supportive diagnostic modalities are lacking, use of tests for biological classifications may be unavailable. Therefore, the role of in-person examination, focusing on the core motor symptoms of parkinsonism and supportive/exclusive criteria as defined by the current clinical criteria for PD and pPD, remains crucial.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by Slovak Research and Development Agency under contract no. APVV-22-0279 and by the Slovak Scientific Grant Agency under contract no. VEGA 1/0712/22 for the Slovak site.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.