
Abstract
While significant progress has been made in treating Parkinson's disease (PD) symptoms, disease-modifying therapies (DMTs) have consistently failed. To address the underlying molecular mechanisms of PD, two biology-based criteria have been proposed: the “Synucleinopathy-Neurodegeneration-Genetics” (SynNeurGe) and “neuronal α-synuclein disease” (NSD) frameworks. Both frameworks emphasize the importance of biological markers over clinical symptoms. They recognize α-synuclein aggregation and genetic mutations (such as SNCA) as key diagnostic elements, with α-synuclein seed amplification assays (SAA) in cerebrospinal fluid (CSF) used to detect early disease stages. Dopaminergic neurodegeneration, measured by DAT imaging, is also central to both frameworks. These shared features aim to improve early diagnosis and precision medicine for PD. However, SynNeurGe provides a broader, more flexible framework that integrates α-synuclein pathology (S), neurodegeneration (N), and genetics (G), linked to clinical features (C). It aims to accommodate the complexity of PD and related Lewy body diseases, facilitating research on targeted DMTs. In contrast, NSD focuses specifically on PD and Lewy body dementia, introducing a staging system (NSD-ISS) based on biological markers and clinical impairment, helping track disease progression from preclinical to symptomatic stages. Despite their differences, both approaches highlight the need for more specific biomarkers and prospective studies to improve early intervention and personalized treatment. Harmonizing SynNeurGe and NSD concepts will be key in creating a universally accepted framework for precise PD diagnosis and therapy development.
Plain language summary
In recent years, significant progress has been made in treating Parkinson's disease (PD) symptoms, but therapies to slow or stop disease progression have failed. Symptomatic treatments focus on neurotransmitter replacement, while disease-modifying therapies target the molecular and cellular causes of PD. Two research criteria aim to redefine PD biologically, not just by clinical symptoms. The SynNeurGe and NSD concepts both focus on the role of α-synuclein, a protein linked to PD, but differ slightly in approach.
The development of a biological classification and staging system for Parkinson's disease (PD) is currently a subject of intense scientific debate. A recent task force from the International Parkinson and Movement Disorder Society (MDS) highlighted the clinical, genetic, mechanistic, and neuropathological heterogeneity of PD, emphasizing the need for a biological definition of the disease. 1 In this invited review, highlighting our recent proposal for a biological classification of PD and related synucleinopathies 2 and contrasting this with another proposal for a biological definition and staging system, 3 we aim to evaluate the proposed concepts, explore the potential for unified classification criteria, and underscore the need for data to address existing knowledge gaps.
The unmet medical need
The last decades have seen significant advances in the development of symptomatic therapies for people living with PD.
4
However, attempts to develop disease-modifying therapies (DMTs) to slow down or stop the progression of the disease have involved a sequence of regrettable failures, with no single therapeutic option being available so far.
4
While symptomatic treatment strategies are mainly based on neurotransmitter-replacement approaches, DMTs aim to interfere with the molecular and cellular mechanisms leading to neuronal dysfunction and degeneration. In recent years, scientific advances have yielded a wealth of insights into the molecular disease mechanisms, thereby identifying appealing targets for therapeutic interventions.4,5 Based on this knowledge, the general concept has emerged that PD is not a homogeneous disease caused by one single and simple biology, but rather a complex condition that can result from different genetic predispositions and environmental triggers, may or may not be associated with pathological aggregation of alpha-synuclein, may affect different parts of the central and peripheral nervous system in different sequential orders, and may evoke a broad spectrum of symptoms in motor, sensory, autonomic, sleep, and cognitive domains.5,6 Specifically, it has been shown, that the molecular mechanisms of PD are active, impairing neuronal function and initiating neurodegeneration many years or decades before the clinical signs and symptoms become apparent.5,6 Therefore, the concept has emerged that a biological rather than a clinical definition of the disease is warranted for a number of reasons:
In this regard, any concept of biological disease characterization may serve one or several of the following purposes:
In contrast to the currently prevailing clinically defined disease concepts of PD,7–9 biological PD criteria may thus be valuable to define the disease entity, for objective and early diagnosis, ideally before symptom onset, for harmonization and stratification of patient groups by active disease mechanisms, for demonstrating presence of targets for specific therapeutic interventions, and possibly for disease staging with the implicit possibility of measuring disease progression.
Two biology-based research criteria for PD have recently been proposed:
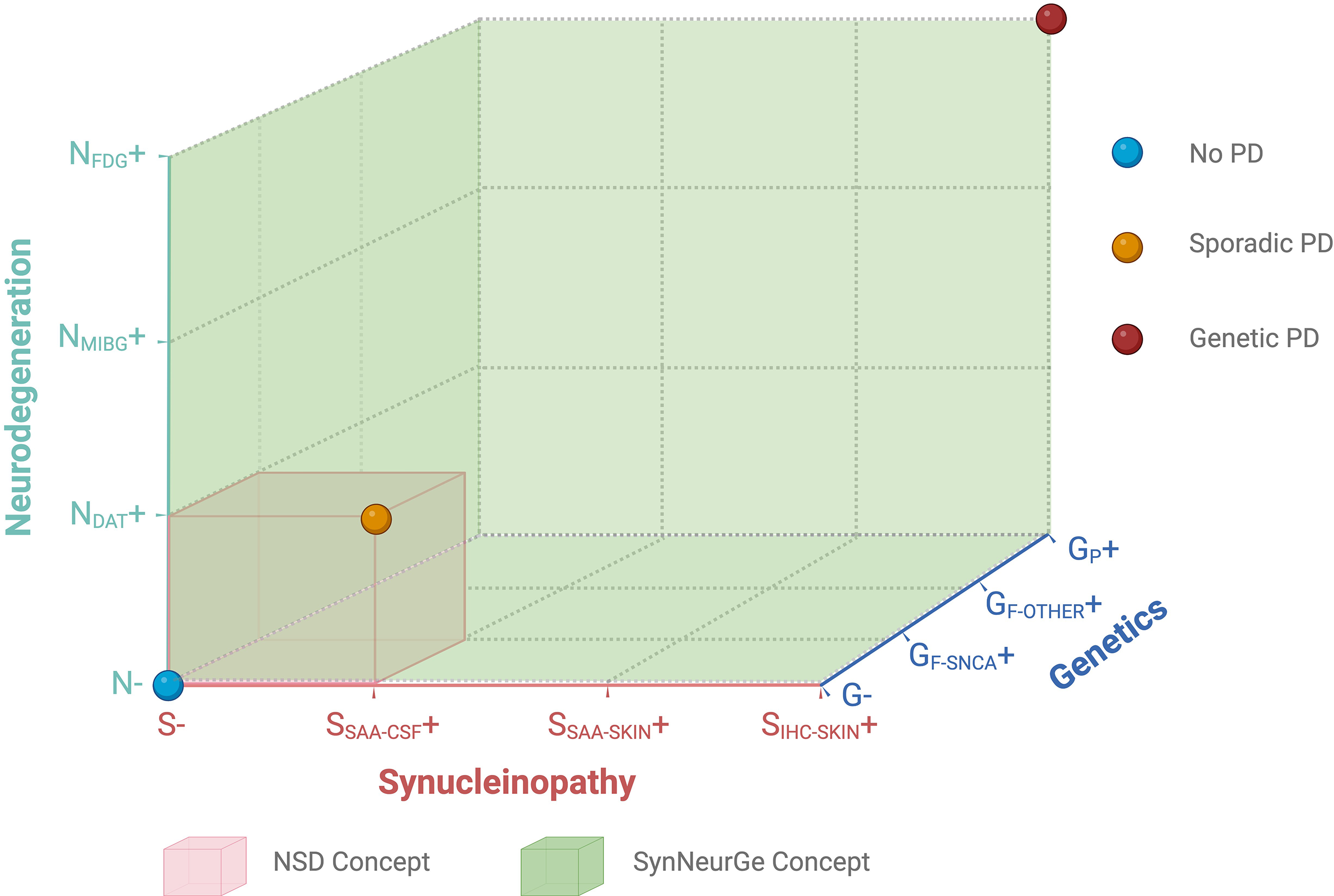
Graphical representation of the overlaps and distinction between the biological SynNeurGe and neuronal synuclein disease (NSD) concepts.
The SynNeurGe and NSD concepts share a number of commonalities. Both consider fully penetrant pathogenic variants in SNCA as sufficient to qualify for a diagnosis of PD. Both endorse α-synuclein SAA in CSF in vivo as sufficient to provide sensitive and quite specific evidence of the presence of pathological α-synuclein aggregates in the central nervous system. Finally, both accept DAT imaging as modality to demonstrate nigrostriatal dopaminergic neurodegeneration associated with PD. As such, the NSD concept essentially defines an entity, which is also embedded as sub-entity within the SyNeurGe classification, that is providing a larger umbrella (Figure 1).
However, the SynNeurGe and NSD concepts display a number of significant differences.
SynNeurGe recommends to use the established terminology of PD as name for the disease (alternatively, DLB where appropriate), and Parkinson's type (or Lewy-type) synucleinopathy as name for prevailing Lewy pathology,
10
whereas the NSD concept introduces the novel terminology “Neuronal Synuclein Disease”, which appears not discernibly distinct from the existing neurobiological concept of “Lewy body disease”
10
and does not reflect the well-known, and probably biologically important presence of astrocytic α-synuclein pathology in PD
11
and of neuronal synuclein pathology in MSA.
12
Additionally, α-synuclein pathology is present in other diseases that are not typically considered ‘primary synucleinopathies’.
1
SynNeurGe proposes a disease concept in which PD involves complex biological processes including both genetic and sporadic forms, which may or may not be associated with α-synuclein pathology (one disease spectrum caused by multifaceted biology), whereas NSD restricts PD to α-synuclein pathology (one disease caused by one biology). SynNeurGe aims to enable a broad spectrum of future research including epidemiology, genetics, neuroimaging, biomarkers, and clinical trials, by proposing a classification system to define subtypes within the broad nosological spectrum of PD. In contrast, NSD is mainly a “biological characterization” conceptualized for therapeutics trials in preclinical or early stages of sporadic PD, not facilitating targeted approaches in genetically determined forms of PD, e.g., with pathogenic variants in PRKN, PINK1, PARK7, LRRK2, VPS35, CHCHD2, or GBA1. SynNeurGe applied the methodology of an evidence- and consensus-based development by academic experts covering the areas of molecular mechanisms, genetics, α-synuclein-pathology, neuroimaging and clinical manifestations of PD, whereas the NSD concept emerged from a consensus process including, amongst others, industry representatives and the US Food and Drug Administration, thereby focussing strongly on drug approval processes. SynNeurGe defines Parkinson's/Lewy type synucleinopathy by prevailing Lewy pathology (Lewy bodies and Lewy neurites), which is neuronal and astrocytic in localisation,11,12 and ultrastructurally characterized by the Lewy-type folding structure of α-synuclein. Parkinson's/Lewy type synucleinopathy is distinct from the multiple system atrophy (MSA) type synucleinopathy, which is oligodendrocytic and neuronal in localisation
12
and displays a different ultrastructural folding structure.
13
In contrast, the NSD concept defines NSD by the presence of pathological (i.e., misfolded and aggregated), neuronal α-synuclein and dopaminergic dysfunction. Since neuronal α-synuclein pathology and dopaminergic dysfunction are also present in MSA, conceptually, the NDS concept does not sharply distinguish between these two very different entities. With respect to the methods used to detect Parkinson's/Lewy type synucleinopathy in vivo, SynNeurGe endorses both CSF and skin SAA as well as skin immunohistochemistry/immunohistofluorescence, emphasizing the need to differentiate MSA by defined exclusion criteria, i.e., additional tests to differentiate both types of synucleinopathy (e.g., NFL in CSF or plasma, structural MRI, MIBG-SPECT, FDG-PET). The background literature and justification for this approach was reviewed in detail in Supplement 1 of the Appendix that accompanied the SynNeurGe paper
2
including Supplementary Panel 1 entitled “Laboratory Differentiation of PD from MSA and other neurodegenerations”. In contrast, NDS proposes CSF SAA only, postulating that MSA can be reliably differentiated by NSD-specific SAA properties. As methods to demonstrate PD-associated neurodegeneration, SynNeurGe endorsed all presynaptic dopaminergic imaging modalities, PD-related metabolic pattern in FDG-PET, and MIBG SPECT to demonstrate peripheral autonomic neurodegeneration (as outlined in detail in Supplement 2 and Supplementary Panel 2 of the Appendix of the SynNeurGe paper
2
), whereas NSD proposes only pre-synaptic nigrostriatal dopaminergic imaging (largely DAT scan). With regard to genetics, SynNeurGe includes both genetic and sporadic PD, stratifies pathogenic gene variants by their penetrance, and acknowledges that several of the classical PD-associated gene variants may not be associated with α-synuclein pathology (i.e., accepting the concept of S− PD) (see also Supplement 3 of the Appendix of the SynNeurGe paper
2
). The NSD concept focusses essentially on sporadic PD, accepting only very rare SNCA variants, thereby excluding most genetic forms of PD. Importantly, SynNeurGe also specifies exclusion criteria for S, N, and G to ascertain specificity of the categories, which is not provided by NSD. Supplement 4 of the SynNeurGe proposal
2
provides a table that outlines a critical appraisal of all of the biological classifications possible in this proposal. SynNeurGe proposed well defined clinical criteria for abnormalities that characterize features possibly and probably related to PD (further explained and characterized in Supplement 5 of the SynNeurGe paper
2
). NSD refers to motor and non-motor signs without characterizing what would constitute sufficient evidence to accept a feature as indicative of underlying neurodegeneration. SynNeurGe defines the disease status as present in patients having 1) a fully penetrant gene variant, 2) N + (regardless of the S status) in G + carriers of pathogenic variants in genes which are compatible with S- PD, or 3) a combination of S + and N + (regardless of G status), whereas an S + status in N- individuals would only be labelled as Parkinson's/Lewy type synucleinopathy. In contrast, the NSD concept labels S + individuals as stage 1A NSD. We believe that the available data on the prevalence of S+ in the aged general population and its predictive value for a later conversion to an actual clinical PD manifestation are completely unknown. Therefore, it is both premature and, indeed, inappropriate to designate these individuals as having a diagnosis of a presently incurable disease (i.e., NSD). The specificity of the α-synuclein SAA has been reported to be within a range of 0.90 to 0.98,
14
which means in turn that 2–10% of individuals not having PD would be mislabeled as having a disease with false positive test results (alternatively this could indicate the presence of pathology (i.e., “incidental Lewy body disease”) that, in the majority, would never become clinically manifest), based on the NSD concept. Unless even more specific tests become available, the SynNeurGe criteria emphasize the need to provide further evidence for N + in addition to S+ before the condition shall be called a disease. SynNeurGe purposefully refrains from proposing disease stages, since we believe that large prospective studies, including individuals initially with no clinical manifestations, are required to prove the temporal order of events in the classification scheme, whereas NSD proposed 7 stages, defined by both biomarkers, clinical manifestation and their impact on daily living. We believe that such large longitudinal studies of asymptomatic S+, N−, G− individuals utilizing advancing research technologies, including developing biomarkers, will be critical to understand the factors that both predispose people to but also protect them from developing clinically manifest Lewy body diseases.
Pros and cons of the two concepts
Both, the SynNeurGe and NSD concepts help to trigger a debate in the scientific community about the need, possibilities, challenges and opportunities to transition the definition of PD and related Lewy body diseases from a traditional, predominantly clinical disease concept to a biology-based disease concept. With the recently gained insights into the molecular underpinnings of PD and the prospect of molecularly targeted therapeutics, we believe it very timely to lead this discussion now, to prepare the PD research and care communities for the development of personalized medicine in PD.
In this context, the NSD concept appears to be less well prepared for future developments, as it tends to view PD from a narrower perspective. While we understand that the research community might accept as convention to restrict the NSD disease concept to a largely sporadic synucleinopathy with dopaminergic deficiency, we argue that this approach is inappropriately restrictive, suggesting a level of knowledge and understanding of disease pathogenesis that is not sufficiently advanced to limit PD to a single biological marker as “definition”. We favor a broader biological classification that incorporates currently available knowledge and can more readily evolve with future scientific advances. Thus, SynNeurGe expands the disease concept to the genetic forms of PD, which have contributed so much to our current understanding of the molecular mechanisms driving the disease and have paved the way for therapeutic developments. Innovative therapeutic approaches such as GBA1-activators, LRRK2-inhibitors, PRKN-activators primarily developed for these genetic forms or PD, are basically out of the scope of the NSD concept. Importantly, the fact that variants in genes such as LRRK2 or PRKN can lead to dopaminergic neurodegeneration and a clinical PD syndrome with or without synucleinopathy imposes intriguing questions as to the relevance of α-synuclein pathology within the pathophysiology of PD. Consequently, the SynNeurGe concept also accepts genetically defined forms of PD that are not accompanied by α-synuclein pathology (i.e., S− forms of PD).
With regard to the methods to demonstrate α-synuclein pathology and neurodegeneration, the NSD concept endorses SAA in CSF and DAT imaging, respectively. While we agree that these methods are suitable for this purpose, again, we believe that the restriction to these two methods is too narrow for the state of the art today, and even more so for the future technologies expected to evolve. In the SynNeurGe concept, we actively endorse α-synuclein-immunhistochemistry (IHC) and SAA in skin as additional tools to demonstrate synuclein pathology in living patients. Indeed, supporting this more inclusive approach, since the publication of the two proposals a large study confirming the utility of IHC has been published. 15 With regard to PD-associated neurodegeneration, we endorse PD-related metabolic pattern in the CNS evidenced by FDG-PET and autonomic denervation of the heart evidenced by MIBG-SPECT as additional tools to demonstrate damage outside of the nigrostriatal dopaminergic system. Both of these approaches are known to be sensitive for PD and even more specific for the differentiation of PD from atypical Parkinson syndromes. Importantly, we emphasize the highly dynamic nature of the field, emphasize the existence of candidate biomarkers in development, and stress the importance of incorporating novel biomarkers as they emerge.
A major distinction between the two concepts is the proposed staging system of the NSD, which incorporates biological events in the preclinical stage, and functional aspects in the clinical stage of the disease. We agree that a biological staging is an essential prerequisite to develop DMTs in preclinical situations. Although we are aware of the efforts being made to validate their staging system, we would emphasize, that the NDS-ISS currently does not appear to be fully supported or validated by existing evidence. To our knowledge, no prospective preclinical data are available to demonstrate its utility in sporadic PD patients in support of the proposed stages of disease progression. We also doubt, whether the two proposed tools (SAA in CSF and DAT-imaging) are sufficient to serve this purpose. Due to the limited available evidence so far, the SynNeurGe concept actively avoided proposing a biological staging.
In the clinical stages, the NSD-ISS follows the principle concept proposed by the ATN approach to Alzheimer's disease 16 and the HD-ISS for Huntington's disease, 17 emphasizing the successive impairment in activities of daily living, however without providing any proposal for its operationalization. Further, a number of important exceptions exist to the implied evolution of the disorder progressing through stages 2A (subtle clinical features without dopaminergic deficit) to 2B (subtle clinical signs with dopaminergic deficit) to 3 (motor or non-motor features causing slight functional impairment, always with dopaminergic deficit). A very substantial group of patients with RBD, DLB, and pure autonomic failure have notable functional impairment due to synucleinopathy, even in the moderate-severe range, yet have normal DAT scan. Presumably this reflects patterns of neurodegeneration in which the dopamine system is affected relatively late. 18 These patients are unclassifiable under the current NSD-ISS, pointing to the troublesome centrality of DAT scan in this concept. Currently available scales to monitor disease progression in clinically established PD patients (e.g., Hoehn & Yahr, UPDRS, SCOPA-COG, NMSS, etc.) are not yet incorporated into this scheme. Also the effect of symptomatic medication on the staging is not specified, i.e., if the staging has to be done in on- or off stage.
The SynNeurGe concept did not propose a staging for clinical PD, but proposed a radically different clinical perspective on PD. We suggest that in persons with biologically defined PD, any possible manifestation associated with the disease, i.e., the broad spectrum of well-defined signs and symptoms in motor, sensory, autonomic, sleep, and cognitive domains, might serve to determine the clinical manifestation(s) of the disease. This enables the field to consider the clinical course of PD in a much broader perspective than it had been possible in the past, where a clinical definition had to be specific enough to allow a diagnosis, before the natural history could be described. Again, as pointed out above, prospective longitudinal data need to be collected to describe the evolution of this clinical spectrum from the starting point of biologically defined disease in the asymptomatic stage.
Both SynNeurGe and NSD are limited by the lack of prospective data that reliably report on the predictive value of the respective S, N and G parameters in healthy individuals for the future development of clinical signs and symptoms of PD. Therefore, many uncertainties regarding the natural history of S+, N + and G + in clinically healthy individuals still need to be clarified before they can be considered as diagnostic parameters that allow clinical decisions to be made on their basis. In order to develop valid and clinically useful diagnostic criteria and staging systems, retrospective analyses of existing cohorts collected in longitudinal studies and, in particular, prospective cohorts to validate and track the development of S+, N + and G + in clinically healthy individuals are required. In particular, very little data is available on the relevance of S+ in N− and G− clinically healthy individuals, which poses major ethical challenges for the communication of such laboratory results. The ethical implications are particularly relevant as long a biomarker-based predictive diagnosis 1st yields diagnostic inaccuracy and 2nd leaves individuals without therapeutic options in absence of approved DMTs.
The road ahead
From the aspects outlined above, it is evident that both the SynNeurGe and NSD approaches are currently research-stage concepts and not ready for application in clinical routine practice. While the NSD approach restricts itself to largely sporadic patients with synucleinopathy and dopaminergic deficit and proposes a staging based on these two parameters, the SynNeurGe approach serves the classification of a broad and complex PD/Lewy body spectrum, based on a more comprehensive toolkit. As outlined above, there are several important areas of missing evidence, which need to be resolved through prospective cohort studies. Specifically, the frequency and latency for conversion of the biological categories (S+, N+, G+, and their respective sub-categories, and combinations thereof) into clinically manifest disease need to be documented in numbers sufficiently large to reliably establish the evolution of biological diagnostic criteria and stages upon which clinical trials can be more effectively planned and conducted. This especially relates to the temporal evolution of the biological anchors defined above in the preclinical stage, in order to enable interventional trials in a presymptomatic or prodromal stage.
The biomarkers that have been proposed as examination methods must be further refined and optimized. For their application in clinically healthy individuals, specificities in the order of 90–98%, 14 as currently valid for SAA in CSF, are still too non-specific, as 2–10% “false-positive” results (with respect to the prediction of manifest disease) are unacceptably high in view of the implications derived from them. Furthermore, the SAA in CSF appears very sensitive, once the cerebral cortex is involved, but less so in incidental Lewy body disease, when Lewy bodies occur in limited regions. 19 Therefore, both sensitivity and specificity of biomarkers for synucleinopathy need to be optimized against the gold standard of brain autopsy. Very likely, additional biomarkers for selected pathways (mitochondrial, lysosomal, inflammatory, etc.) and biomarkers for co-pathologies (e.g., ATN, vascular, inflammatory co-pathologies) and other mitigating factors (e.g., exercise, education) will become important in the future to accommodate for individual deviations from predicted disease trajectories. Possibly one of the greatest unmet needs for any biological approach to PD and related Lewy body diseases is the development of more sensitive and reliable markers of the multisystems neuronal dysfunction and degeneration (i.e., not simply dopaminergic) that can serve as both state and progression markers at all stages of the disease. Also, the clinical definitions for the disease manifestation, as operationalized in the SynNeurGe criteria, require prospective validation and longitudinal natural history data. In the NSD concept, the ADL impairment requires operationalization in order to become useful. Therefore, immediate next steps for the research community will be the generation and analysis of longitudinal cohorts, the validation and longitudinal documentation of the change in biomarkers over time, and the incorporation of novel biomarkers as they emerge. In view of the controversial debate in the scientific community about the paradigm shift from a predominantly clinical to a biological concept of disease, stakeholders at large as well as patients and caregivers need to be involved in a debate about the prevailing future concepts of disease. One important goal of the public debate will be the harmonization of the SynNeurGe and NSD concepts to arrive at an approach that can be universally accepted in the community. Furthermore, both proposed concepts are currently technology-heavy approaches and might disfavor low- and middle-income countries. Therefore, future developments will also have to consider low-budget, easy to handle alternative biomarkers.
In the long term, biological criteria for disease definition, diagnosis, target demonstration and engagement, staging, and progression measurement will become broadly used and will pave the way towards individualized precision medicine approaches in PD.
Footnotes
Acknowledgments
We would like to thank Charles H. Adler, Daniela Berg, Christine Klein, Tiago F. Outeiro, Werner Poewe, Ronald Postuma and A. Jon Stoessl for their excellent collaboration in the development of the SynNeurGe criteria. We thank Thomas Koeglsperger for help with the illustration.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Günter Höglinger was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198).
Anthony E. Lang received grants from Brain Canada, Canadian Institutes of Health Research, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, Parkinson Foundation, Parkinson Canada, and W. Garfield Weston Foundation.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Günter U. Höglinger has ongoing research collaborations with Roche, UCB, Abbvie; serves as a consultant for Abbvie, Alzprotect, Amylyx, Aprinoia, Asceneuron, Bayer, Bial, Biogen, Biohaven, Ferrer, Lundbeck, Novartis, Roche, Sanofi, Servier, Takeda, Teva, UCB; received honoraria for scientific presentations from Abbvie, Bayer, Bial, Biogen, Bristol Myers Squibb, Kyowa Kirin, Pfizer, Roche, Teva, UCB, Zambon; received publication royalties from Academic Press, Kohlhammer, and Thieme.
Anthony E. Lang has served as an advisor for AbbVie, Amylyx, Aprinoia, Biogen, BioAdvance, Biohaven, BioVie, BlueRock, BMS, Denali, Janssen, Lilly, Pharma 2B, Sun Pharma, and UCB; received honoraria from Sun Pharma, AbbVie and Sunovion; is serving as an expert witness in litigation related to paraquat and Parkinson's disease, received publishing royalties from Elsevier, Saunders, Wiley-Blackwell, Johns Hopkins Press, and Cambridge University Press.