
Abstract
There is an estimated 35–45% loss of striatal dopamine at the time of diagnosis of Parkinson’s disease (PD), and cases clinically diagnosed in the early stages may already be pathologically in advanced stages. Recent large-scale clinical trials of disease-modifying therapies (DMT) also suggest the necessity of targeting patients at earlier stages of the disease. From this perspective, the prodromal phase of PD is currently the focus of attention, emphasizing the need for a prodromal mouse model that accurately reflects the pathophysiology, along with early biomarkers. To establish prodromal animal model of PD with high face validity that reflects the disease state, the model must possess high construct validity that accurately incorporates clinical and pathological features in the prodromal phase. Furthermore, as a preclinical model of DMT, the model must possess high predictive validity to accurately evaluate the response to intervention. This review provides an overview of animal models which reflect the characteristics of prodromal PD, including alpha-synuclein (aS) accumulation and associated early non-motor symptoms, with a focus on the aS propagation model and genetic model. In addition, we discuss the challenges associated with these models. The genetic model often fails to induce motor symptoms, while aS propagation models skip the crucial step of initial aS aggregate formation, thereby not fully replicating the entire natural course of the disease. Identifying factors that induce the transition from prodromal to symptomatic phase is important as a preclinical model for DMT to prevent or delay the onset of the disease.
Keywords
INTRODUCTION
In Parkinson’s disease (PD), striatal dopamine loss at diagnosis is estimated to be 35–45% [1], and in many cases, Lewy pathology (LP) is assumed to have progressed to stage 3 or higher in the Braak stage. Even in cases clinically diagnosed as early-stage there is a possibility that pathologically the disease is already in an advanced stage when considering the entire natural history of PD. Recent results from large-scale clinical trials of disease-modifying therapies (DMT) [2–4] suggest the need to reconsider the pathogenesis of the disease and to target patients with earlier stages of disease [5]. From the viewpoint of therapeutic development, the prodromal phase of PD is currently the focus of attention, and a mouse model in this prodromal phase that accurately reflects the pathophysiology as well as early biomarkers for this phase is highly needed.
VALIDITIES NECESSARY FOR PRODROMAL PD MODELS
Animal models of disease are usually required to have three types of validity: construct validity, face validity, and predictive validity [6]. Construct validity assesses how well the model reflects the known pathophysiological mechanisms in humans, face validity evaluates how closely the created model replicates the human phenotype, and predictive validity assesses the model’s ability to predict responses to interventions or pathophysiology in humans. To create an animal model with high face validity reflecting the pathology, it is essential to accurately understand the clinical symptoms and pathological findings during the prodromal phase and confer the model with high construct validity.
Clinical aspects for prodromal PD
A variety of non-motor symptoms (NMS) manifest during the prodromal period of PD, many of which have been present for several years or even more than a decade, providing a therapeutic window for DMT. Of particular significance are those that exhibit a high predictive value for the future development of PD. Polysomnography (PSG)-proven REM sleep behavior disorder (RBD) is the most important NMS with the positive likelihood ration (LR+) of 130, followed by orthostatic hypotension (OH) (LR+: 18.5), hyposmia (LR+: 6.4), and constipation (LR+: 2.5) [7].
The neuroanatomical substrates for these prodromal symptoms also provide important information about the initial lesions in PD. For example, the locus subceruleus and ventral medulla are thought to be responsible for RBD [8], the peripheral sympathetic nervous system (SNS) for OH, and the peripheral parasympathetic nervous system for constipation. Regarding hyposmia, in addition to the olfactory system including the amygdala and the piriform cortex, the cholinergic system is also implicated in hyposmia [9]. Importantly, multiple prodromal symptoms often occur in combination [10]. Among these combinations, the presence of NMS combinations in which the responsible lesions are distant and seemingly unconnected such as constipation and hyposmia is important in considering the pathophysiology of prodromal PD [11].
Pathological aspects for prodromal PD
To understand the pathological changes occurring in the prodromal phase of PD, pathological studies using consecutive autopsied brains are essential, rather than case-control studies in PD. In their classic paper in 2003, Braak et al. proposed a hypothesis that in typical PD cases, the earliest lesions appear in the dorsal motor nucleus of the vagus nerve (DMV) and the olfactory bulb (OB) with subsequent LP predominantly ascending in the brainstem [12]. In 2009, Beach et al. analyzed cases including concomitant Alzheimer’s disease (AD) or dementia with Lewy bodies (DLB), and including the OB in their analysis. They argued that the earliest lesions are most common in the OB [13]. The continuous progression observed in these studies implies a prion-like spread of alpha-synuclein (aS). In contrast, in 2021, Tanei et al. conducted an analysis, including the OB and peripheral tissues, and reported that besides the DMV and OB, the SNS also showed early lesions [14]. In this study, among 178 LP-positive samples from 518 consecutive autopsies, cases with multifocal LP in poorly interconnected areas, despite being in the early stage, were described. This suggests the possibility that not all cases can be explained by the continuity of progression through prion-like spread, and there are cases where the lesions initiate multifocally.
ANIMAL MODELS OF PRODROMAL PD
For a model of prodromal PD, it is ideal to exhibit multiple prodromal symptoms with high predictive value due to aS pathology, followed by the manifestation of motor symptoms associated with the progression of aS pathology. Furthermore, as a preclinical model for DMT, high predictive validity is also required to accurately evaluate the response to intervention. In this section, we focus on models that reflect the prodromal phase of sporadic PD, i.e., models that show accumulation of aS and associated prodromal symptoms. It has been reported that LP does not always correlate with cell death or neurological symptoms in human PD [15], and that dopamine (DA) dysfunction can cause gastrointestinal (GI) or olfactory dysfunction in mice [16, 17]. However, in most cases, the neuronal dysfunction due to the accumulation of aS, regardless of its fibrillar or oligomeric form, is considered to be responsible for prodromal NMS. Based on the clinical symptoms and pathological findings of prodromal PD mentioned earlier, this section provides an overview, categorizing the models into the aS propagation model and genetic model. In addition, in classical toxin-based models, the prodromal model is gaining attention, and some of them are being introduced.
Propagation models
The propagation of aS aggregates in the mouse brain through the administration of aS preformed fibrils (PFF) was first demonstrated by Luk et al. in the striatum [18]. The strategy of administering aS PFF to the earliest lesions of LP inferred from pathological studies, is considered reasonable for reproducing the entire course of PD, including the prodromal stage (Table 1).
Prodromal symptoms in aS propagation and genetic models
aS: alpha synuclein, BAC: bacterial artificial chromosome, CSF: cerebrospinal fluid, DMV: dorsal motor nucleus of the vagus nerve, GI: gastrointestinal, LC: locus coeruleus, mo: month old, mpi: months post-inoculation, ND: not described, OB: olfactory bulb, PAC: P1-artificial chromosome, PFF: preformed fibril, RN: raphe nucleus, RWA: REM without atonia, SLD: sublateral dorsal nucleus, SNpc: substantia nigra pars compacta, SNr: Substantia nigra pars reticulata, wpi: week post-inoculation,+/+: homozygous,+/– : heterozygous.
Olfactory bulb models
aS aggregates administrated into the OB of wild-type mice has been reported to rapidly propagate to the olfactory and limbic systems, as well as some brainstem regions, without exhibiting symptoms other than olfactory impairment [19]. However, when aS transgenic (Tg) mice were employed in this experiment, they exhibited symptoms of cognitive decline and increased anxiety [20]. These may be considered DLB-like conditions, but the pathology is atypical for DLB in that there is no progression to the cerebral cortex, and they did not exhibit the mixed pathology of AD commonly observed in most of human DLB cases. These models also show a regression of aS pathology with age, probably due to neuronal cell death [20, 21]. Additionally, attempts were made to administer aS PFF to the olfactory mucosa of wild-type mice. This was based on the assumption of potential entry of neurotoxic substrate from the external environment, taking into consideration the pathological evidence of aS deposition in the olfactory epithelium of individuals with PD [22]. However, there was no progression of aS pathology beyond the OB into the brain [23]. Recently, surprising results have been reported from a study involving the intranasal administration of exosomes derived from cerebrospinal fluid of PD patients to wild-type mice. The findings include a decrease in motor performance in the rotarod test and increased anxiety in the open field test three months later, hyposmia four months later, and DA neuronal loss eight months later [24].
Gastrointestinal models
One of the earliest lesions in PD is identified in the DMV. However, due to the technical difficulty in administering aS PFF to the DMV and the assumption that the GI tract, which has connections with the DMV, is considered the origin of LP, aS PFF administration is performed in the GI tract. In early reports using wild-type mice, rats, and primates, the spread was confined to the DMV or LC and no further upward propagation was observed [25–28]. However, in aS Tg rats, propagation to the substantia nigra pars reticulata was reported [29], and in adult rats, spread to the OB was documented [30]. In the latter model, extensive pathology was observed in the SNS and the pathway from the SNS to the LC via the spinal cord is assumed to be the route of propagation to the central nervous system. However, the involvement of vagal propagation is unknown because no vagotomy experiments have been performed.
The most extensive propagation model among these was reported by Kim et al. (2019) [31]. In this model, aS PFF were injected into the stomach and duodenum of young wild-type mice. LP propagated throughout the brain to the OB, accompanied by NMS such as decreased Gl motility and olfactory impairment. Additionally, the model exhibited DA cell death and motor impairments. Moreover, these phenotypes were completely abolished by vagotomy. The success of the GI administration model suggests the potential to modulate the pathology from the peripheral, including the GI tract and gut microbiota, opening possibilities for DMT. However, the reproducibility between laboratories remains a significant challenge. Variability in results is known to occur due to factors such as the production and storage methods, sonication, and administration sites of aS PFF. Therefore, standardization or optimization of aS administration protocols is crucial for future research [32].
Autonomic nervous system model
Recently, the autonomic nervous system, especially the SNS, has attracted attention as one of the earliest lesions in PD. When aS PFF was administered to sympathetic ganglia in heterozygous (+/–) M83 aS Tg mice, which express A53T mutant human aS under the murine prion promoter, the fibrils propagated to peripheral sympathetic tissues by antegrade transmission and to brainstem nuclei such as periaqueductal gray, raphe nuclei and LC by retrograde transmission [33]. As a result, the mice exhibited severe autonomic dysfunction, manifesting as orthostatic hypotension, constipation, and hypohidrosis, without motor dysfunction. This mouse model is considered to resemble pure autonomic failure. However, no propagation was observed in wild-type mice, suggesting that the phenotype may depend on the site of aS overexpression in M83 + /– mice, which have high transgene expression in the spinal cord.
RBD-related region (SLD) model
The sublaterodorsal nucleus (SLD) and ventromedial medulla have been identified as responsible regions for RBD in mice. SLD provides excitatory output to ventromedial medulla neurons, which send inhibitory output to motor neurons in the spinal cord. These neurons are REM-on neurons, meaning they are activated during REM sleep, and thus their dysfunction can cause RBD [34].
A very low dose (1μg) of aS PFF administered to the SLD led to REM without atonia, an electrophysiological feature of RBD, as observed in PSG 1-month post-inoculation (mpi) [35]. At 3 mpi motor symptoms accompanied by decreased number of TH-positive cells in the substantia nigra pars compacta and DA content in the striatum were observed. At 5 mpi, NMS such as GI dysfunction and hyposmia were manifested. The fact that motor symptoms preceded NMS other than REM without atonia, and the lack of pathological evidence that SLD is an initial lesion in PD, raises concerns about face and construct validity. However, the administration of a very small amount of aS PFF to brainstem nuclei with extensive connections to various regions of the brain resulted in widespread propagation of aS and various PD-related symptoms, including prodromal symptoms. This aspect is important for considering the mechanism of the disease progression.
Genetic models
A limited number of models have been developed to simulate familial PD with pathological features such as Lewy bodies (LB) and NMS as prodromal manifestations, similar to sporadic PD. Among these, the most extensively reported models are aS Tg models, followed by leucine-rich repeat kinase 2 (LRRK2) models. The common feature of these two genes is that they serve as causative genes for familial PD and risk genes for sporadic PD (Table 1).
aS Tg models
There are several reports that aS expression is upregulated in PD brains [36]. Additionally, there have been findings that risk single nucleotide polymorphisms identified in genome-wide association studies and risk polymorphisms in the Rep1 region are associated with increased expression of aS [37, 38]. Furthermore, given that the duplication of aS is implicated as a cause of familial PD closely resembling sporadic PD [39], aS Tg mice have construct validity if the expression levels of aS are appropriate.
The phenotype of aS Tg models depends on the site and level of aS expression. Historically, mice have been generated using high-expression promoters such as platelet-derived growth factor, Thy-1, and mouse prion, contributing significantly to the elucidation of aS toxicity and the development of DMT targeting aS. However, models with ectopic high expression of aS often exhibit phenotypes that are not relevant to human PD [40]. Nevertheless, mice displaying NMS associated with aS aggregation have also been reported. For example, Thy-1 aS Tg mice exhibit deficits in olfaction, GI dysfunction, sleep alterations, cognitive dysfunction, anxiety phenotype, and other NMS before the onset of DA loss at 14 months [41–44 and reviewed in 45], and aS Tg mice driven by the prion promoter also exhibit deficits in olfaction and GI dysfunction [46–51].
In families with aS gene multiplication, the presentation of prodromal NMS such as RBD and autonomic dysfunction is well-documented [39]. Particularly in duplication families, there is clinical and pathological resemblance to sporadic PD [39]. To replicate the native expression pattern of aS and model familial PD with duplications or triplications of aS, aS P1 artificial chromosome (PAC) or bacterial artificial chromosome (BAC) Tg mice have been created.
In humanized mutant (A53T) aS PAC Tg mice harboring SNCA knock-out allele, deposition of proteinase K-resistant aS in the GI tract was accompanied by a decline in colonic motility, as evidenced by an extended whole-gut transit time by three months of age [52]. This mouse model exhibited mild motor symptoms without aS pathology, and thus may demonstrate abnormalities in the GI tract in the very early stages of PD. It has been reported that administration of Posiphen, which decreases human aS transcription, improved colonic motility in these mice [53].
Another genome-based aS Tg mouse model, the A53T aS BAC Tg mouse, exhibited prodromal symptoms such as hyposmia and RBD by 9 months of age [54], prolonged whole gut transit time (data not shown in [54]) and about 17% DA cell loss by 18 months, although it did not show motor symptoms [54]. Furthermore, in these mice deposits of phosphorylated and oligomeric aS were found in vulnerable regions of PD, including the OB, GI tract, DMV, substantia nigra pars compacta, and regions responsible for RBD, such as the SLD and the ventral medulla [54]. Although no clear aS fibril formation was observed in this mouse, and the presence or absence of aS propagation is not known, it is noteworthy that only mild expression of aS at the native expression site can cause multifocal aS foci and multiple NMS, representing a multifocal foci model [55].
Wild-type aS BAC Tg rats demonstrated age-dependent accumulation of aS, exhibiting olfactory impairment at 3 months, motor dysfunction at 12 months, and approximately 39% DA cell death by 18 months [56]. However, wild-type aS BAC Tg mice alone, utilizing the same BAC Tg construct as BAC Tg rats, showed hyperlocomotion but did not show DA cell loss [57, 58]. This suggests a potential resistance of mouse DA neurons to aS toxicity compared to rats. Similar to the observations in PAC Tg mice, where no phenotype was observed in the wild-type aS Tg mice, the presence of the A53T mutation, known to enhance aS aggregation, is hypothesized to contribute to the phenotypic enhancement observed in A53T aS BAC Tg mice.
LRRK2 models
LRRK2 is not only a causative gene for familial PD but also a significant risk gene for sporadic PD [59]. In familial PD with LRRK2 mutations, the correlation between the genotype and clinical/pathological phenotype is complex, and it is known that even within the same family, these correlations can vary [60]. Therefore, not all patients exhibit the same prodromal symptoms as sporadic PD. However, there are reports suggesting an association between the presence of LP and NMS such as anxiety, orthostatic hypotension, and cognitive decline in familial PD with LRRK2 mutations [61]. Additionally, LRRK2 mutations are causative genes for autosomal dominant inherited PD, and studies have reported only a mild increase in the expression levels of LRRK2 protein in incidental LB disease brains, with no significant increase in PD brains [62]. From the viewpoint of construct validity, knock-in (KI) models may have advantages over Tg models.
R1441C KI mice exhibit hyposmia at 24 months of age, but no obvious aS accumulation or motor symptoms [63]. R1441G KI mice have been reported to show increased aS oligomers in the cortex and striatum at 18 months of age [64]. G2019S KI mice show a higher prevalence of sleep fragmentation, similar to sporadic PD, at 8–10 months [65]. However, overall, the manifestation of NMS is mild, and the formation of aS aggregates is often not observed. From the perspective of face validity, these models may not be considered sufficient as prodromal PD models.
VMAT2-deficient model
Monoaminergic neurons, such as DA and noradrenaline (NA) neurons in brains and NA neurons in the SNS, are highly vulnerable in PD. VMAT2 plays a role in packaging monoamines into synaptic vesicles, protecting nerve terminals from oxidative stress generated by free cytosolic monoamines [66]. VMAT2-deficient mice which express ∼5% of normal VMAT2 (VMAT2 LO mice) demonstrate aS accumulation and age-dependent DA and NA, but not serotonergic degeneration [67, 68]. Furthermore, these mice also exhibit NMS such as progressive hyposmia, delayed gastric emptying, altered sleep latency, anxiety-like behavior, and depressive behavior [69]. This suggests that dysfunction of monoaminergic neurons contributes to various prodromal NMS in PD, and VMAT2-deficient mice may serve as a useful prodromal PD model.
Toxin-based models
Classical toxin-based models, especially the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration model in non-human primates, have made significant contributions as preclinical models for clinical trials targeting motor symptoms over the years. Currently, attention is also being directed towards prodromal symptoms in these models. For instance, MPTP-administered marmosets exhibit sleep disturbance and altered circadian rhythm [70], while MPTP-administered macaques display a disorganization of nighttime sleep with reduced deep sleep quality and excessive daytime sleepiness [71]. Regarding toxin-based rodent models, readers are referred to the following reviews [55, 72].
The advantages of these toxin-based models lie in inducing clear motor symptoms. However, drawbacks include the short duration of the prodromal phase before the manifestation of motor symptoms, especially in acute models. Additionally, many of these models do not exhibit aS aggregates, and their construct validity is also considered low.
CHALLENGES AND FUTURE DIRECTIONS
Challenges as a preclinical model
Animal models have successfully replicated multiple prodromal symptoms. The aS PFF injection model represents the aS propagation hypothesis, while the aS genetic model represents the multifocal initiation model [55]. In human PD, there is a possibility of propagation occurring from a multifocal initiation site, as represented by these combinations. The genetic model focuses on the onset of the disease but mostly fails to induce motor symptoms. Some aS propagation models exhibit robust propagation and DA cell loss, yet they skip the formation of the most crucial early-stage aS aggregate formation. Thus, there is a gap between these two models, and the entire natural course of the disease, from the prodromal to the symptomatic phase, is not fully replicated (Fig. 1). Reproducing the process from aggregation to propagation and replicating the transition between these two phases is considered crucial as a preclinical model for DMTs aimed at preventing or delaying onset of PD.
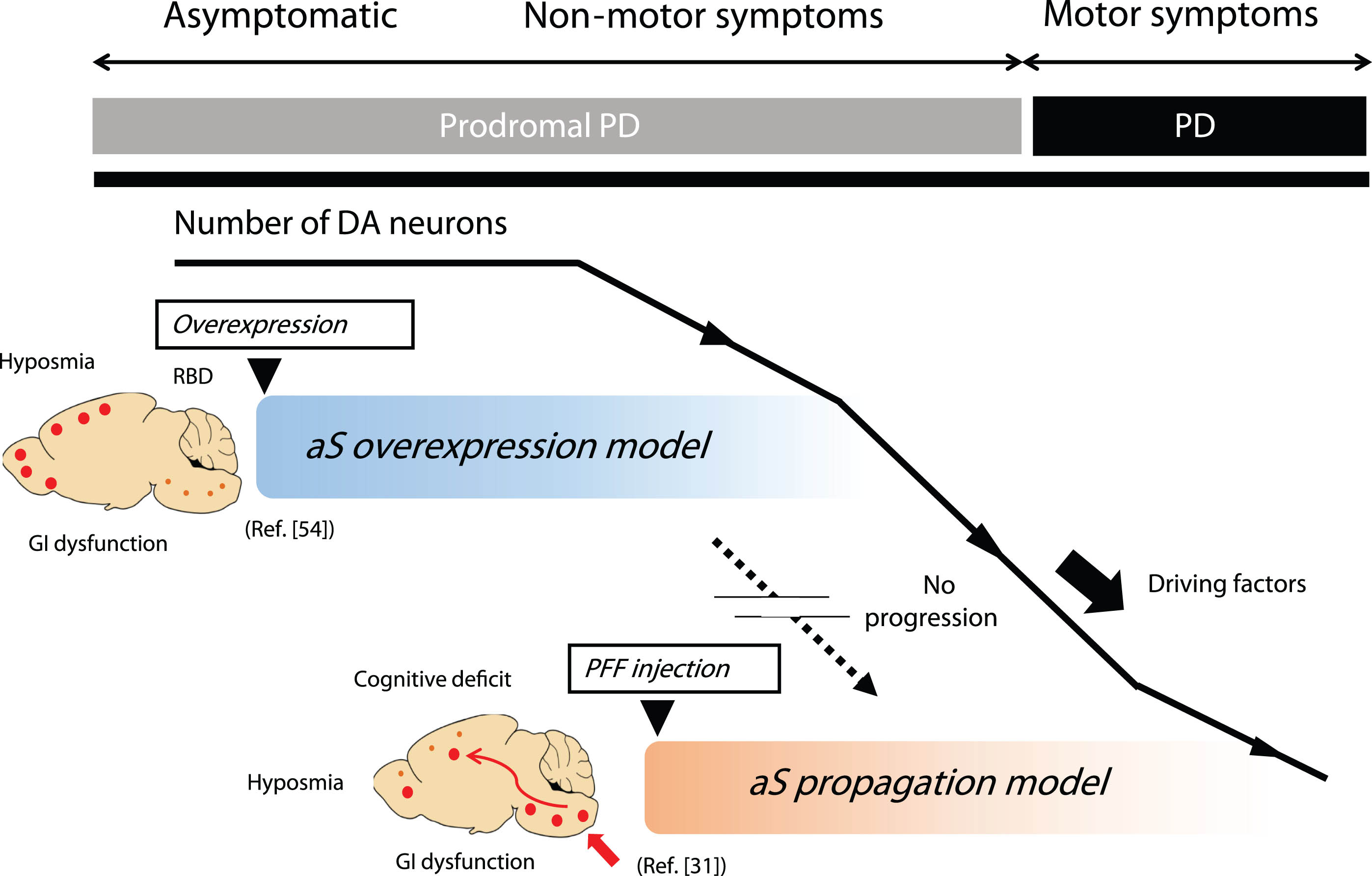
Representative animal models of prodromal PD. The genetic model represented by the aS overexpression model focuses on disease onset but fails to show motor symptoms associated with DA cell loss. While most successful aS propagation models exhibit robust propagation and DA cell loss, they often skip the critical initial step of aS aggregate formation. Thus, the entire natural course of the disease, from the prodromal to the symptomatic phase, is not fully replicated by a single animal model. This limitation is one of the drawbacks as a preclinical model for DMT, targeting the inhibition of progression from the prodromal to symptomatic phase of PD. aS, alpha-synuclein; DA, dopamine; GI, gastrointestinal; PD, Parkinson’s disease; PFF, preformed fibrils; RBD, REM sleep behavior disorder.
What environmental or genetic factors contribute to the formation of aS aggregates with high seeding activity?
Regarding the seeding activity and propagation of aggregated aS in aS Tg mice, it has been reported that brain lysate exhibits seed activity, especially in aS Tg models with high levels of aS expression [73, 74], although this seeding activity may be lower compared to that in human DLB brains [74]. However, the existence of propagation in vivo in aS Tg mice has not been conclusively demonstrated [75, 76]. Sastry et al. reported the absence of propagation using a tetracycline-inducible system with multiple aS Tg mice [77]. On the other hand, mice with above-mentioned high seeding activity in brain lysate typically utilize a high-expression system with a pan-neuronal promoter, making it practically difficult to evaluate the in vivo propagation to non-aS expressing neuronal cells.
Viral vector-mediated overexpression systems have been reported to induce the transfer of aS [78]. However, it is crucial to clearly distinguish between transfer or diffusion and the ‘propagation’ which is caused by a self-templating mechanism involving endogenous aS. Indeed, in the above-mentioned aS overexpression model by adeno-associated virus vector, enhanced aS pathology was observed in aS knockout mice, suggesting evidence of transfer rather than propagation of aS [79]. Considering the presence of LP in approximately 34% of consecutive autopsy brains [14], understanding the cellular and extracellular environmental factors that confer high seeding and propagation activity to aS is a critical challenge.
Once aS acquires seeding activity and intracellular aggregation initiates, it is hypothesized to induce cellular damage by involving membrane structures of intracellular organelles, such as mitochondria, regardless of forming typical LB composed of fibrillar aS [80]. However, in vivo, it remains a challenge to determine whether aggregated aS is confined within cells as LB, or if it is transferred to neighboring cells in the form of oligomers or small fibrils, leading to further propagation. This aspect requires further investigation. Factors conferring strong seeding activity to aS may serve as crucial targets for DMT, potentially acting as progression factors from the prodromal phase to the symptomatic phase.
CONCLUSION
Various early biomarkers and diverse drug discovery seeds have been developed, and the time is ripe for the development of DMT for PD. Models of prodromal PD are not only essential as preclinical models for DMT but also contribute to a deeper understanding of the pathophysiology of prodromal PD which is limited in human studies. The development of models closer to human PD, also considering individualization and stratification, is desired.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This study was supported by grants from the Japan Science and Technology Agency (JST) Moonshot Research and Development Program (HY and RT, no. JPMJMS2024)
CONFLICT OF INTEREST
The authors have no conflict of interest to report.