
Abstract
Parkinson’s disease (PD) is an increasingly common neurodegenerative disease. It has been suggested that the etiology of idiopathic PD is complex and multifactorial involving environmental contributions, such as viral or bacterial infections and microbial dysbiosis, in genetically predisposed individuals. With advances in our understanding of the gut-brain axis, there is increasing evidence that the intestinal microbiota and the mammalian immune system functionally interact. Recent findings suggest that a shift in the gut microbiome to a pro-inflammatory phenotype may play a role in PD onset and progression. While there are links between gut bacteria, inflammation, and PD, the bacterial products involved and how they traverse the gut lumen and distribute systemically to trigger inflammation are ill-defined. Mechanisms emerging in other research fields point to a role for small, inherently stable vesicles released by Gram-negative bacteria, called outer membrane vesicles in disease pathogenesis. These vesicles facilitate communication between bacteria and the host and can shuttle bacterial toxins and virulence factors around the body to elicit an immune response in local and distant organs. In this perspective article, we hypothesize a role for bacterial outer membrane vesicles in PD pathogenesis. We present evidence suggesting that these outer membrane vesicles specifically from Gram-negative bacteria could potentially contribute to PD by traversing the gut lumen to trigger local, systemic, and neuroinflammation. This perspective aims to facilitate a discussion on outer membrane vesicles in PD and encourage research in the area, with the goal of developing strategies for the prevention and treatment of the disease.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is a multifactorial disease encompassing many body systems but is clinically recognized as a debilitating neurodegenerative disease that affects motor function [1]. The cardinal motor symptoms include tremor, bradykinesia, postural instability, and rigidity. Concurrently, there are significant non-motor symptoms, including constipation, loss of a sense of smell, and sleep disturbances [1]. There is no single definitive cause for idiopathic PD (∼90% of cases), instead, it is hypothesized that a mosaic of genetic and environmental risk factors coalesces to initiate disease [2]. These factors include, but are not limited to, exposure to environmental insults (such as neurotoxins, infection, and pollutants) and genetic susceptibility [3–7]. Ultimately, the consequences of these exposures accumulate in the aging body and are suggested to lead to neuroinflammation, the initial loss of synapses in the striatum, neuronal loss in the substantia nigra (SN), and the aggregation of insoluble alpha-synuclein (α-syn) in Lewy bodies, the pathological hallmark features of PD [8].
A conceptual framework has been proposed to characterize the contribution of known environmental and biological factors to disease pathogenesis [8]. This framework proposes there are three phases to consider: triggers, facilitators, and aggravators [8]. Triggers (such as bacterial infections or exposure to industrial toxins) enable disease initiation when present alongside other facilitators [8–10]. Facilitators (such as peripheral inflammation, aging, or genetic susceptibility) are proposed to spread the disease and impact the central nervous system (CNS) [8, 11]. Aggravators (such as neuroinflammation, oxidative stress, and impaired autophagy) are hypothesized to exacerbate and accelerate neuronal dysfunction and /or further the spread of already initiated pathological events [8, 12]. This framework incorporates the multifactorial nature of PD and proposes a continuum of stages whereby factors may differentially contribute to disease progression depending on the disease stage. Inflammation is a consistent feature throughout this framework [8, 13]. The gastrointestinal (GI) tract is considered a site of inflammation in PD [10, 14] with elevated levels of pro-inflammatory bacteria and reduced levels of certain anti-inflammatory bacteria observed in people with PD (PwP) [15]. Researchers have hypothesized that this dysbiosis contributes to the development of disease, however, the mechanism is unclear [10, 17].
Here, we propose outer membrane vesicles (OMVs) released by gut bacteria as facilitators and aggravators of PD. We suggest they are a key mechanism by which gut bacteria mediate intercellular communication and transfer of immunomodulatory compounds around the body, contributing to systemic and neuroinflammation, and consequently neurodegeneration.
INFLAMMATION AND ETIOLOGY OF PD
The etiological risk factors of PD are varied, but many converge in their involvement in inflammatory processes [13, 18]. In the brain reactive microgliosis and elevation of inflammatory markers are some of the most commonly described features alongside neurodegeneration and α-syn deposition [19–26]. Previously, neuroinflammation was explained away as a mere consequence of neurodegenerative processes; however, many research groups have demonstrated in animal models that inflammation itself can contribute to furthering disease processes especially when it becomes chronic [27, 28]. Compellingly proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6, and important inflammatory pathway proteins like nuclear factor-kappa B (NF-κB) and inducible nitric oxide synthase (iNOS) are found to be elevated in the periphery and CNS of PwP [21, 29–31]. Similar elevations of cytokines, other inflammation-associated proteins, and immune cell changes have also been recapitulated in Parkinsonian animal models [32–36]. A role in promoting inflammation is a commonality between many of the proposed etiological factors that are hypothesized to trigger or facilitate PD.
Advanced age is the greatest risk factor for PD, and during aging there is a dysregulation of inflammatory processes due to the life-long cumulative effects of immune activation from various self- and non-self-stimuli [11]. This can lead to the development of sterile, low-grade, and chronic inflammation as a person ages, which is delineated from normal physiological inflammation and termed inflammaging [37, 38]. Many of the common changes to immune function that occur with age overlap with PD-associated immune dysfunction, such as the development of gut dysbiosis, reduced macrophage activity, increased inflammasome expression, and increased blood-brain-barrier (BBB) permeability [38–40].
Genetic variations that are associated with PD are also known to regulate immune responses. Notably, the leucine-rich repeat kinase 2 (LRRK2) gene encodes a protein that is highly expressed in immune cells and is known to have a role in response to bacterial infections and the innate immune system [41, 42]. Mutations in LRRK2 are suggested to lead to chronic overactivation of microglia and neuroinflammation [43, 44]. The gene SCNA encodes for α-syn, a major component of Lewy bodies, and while the physiological roles of α-syn remain unclear, data is emerging that describes a role in immune regulation due to antimicrobial, antifungal, and antiviral effects [6, 45]. PINK1, a protein kinase, and parkin, an E3 ubiquitin ligase, control the elimination of dysfunctional mitochondria from the cell in a process known as mitophagy. PINK1 and PRKN (the genes encoding PINK1 and parkin, respectively) deficient mice show minimal signs of neurodegeneration unless exposed to a pro-inflammatory trigger such as bacterial infection or lipopolysaccharide (LPS) administration suggesting PINK1 and PRKNs roles in protecting against inflammation-driven neurodegeneration [46, 47]. Both DJ-1 and ATP13A2 deficiency attenuate the anti-inflammatory effects of astrocytes [48, 49]. This suggests that these genetic changes may contribute to PD by increasing one’s susceptibility to pro-inflammatory stimuli. Furthermore, over 90 independent risk signals have been identified in recent genome-wide association studies, of which the risk factor gene’s functions are enriched in chemical signaling pathways involved with responding to stressors [50].
Environmental exposure to toxins, mainly pesticides, has been long linked with PD, with a meta-analysis finding a 66% increased risk of developing PD after pesticide exposure [51]. Pesticides like paraquat and rotenone, have direct toxic effects by interfering with mitochondrial function, but have also been shown to activate inflammatory pathways, like cytokine release and microgliosis [52, 53].
Conversely, smoking (nicotine), caffeine consumption, and the regular use of anti-inflammatory analgesics have been proposed to confer protection against the development of PD due to their anti-inflammatory effects [54–58].
Bacterial, viral, and fungal infections are associated with an increased risk of PD [10, 60]. Specific examples of these infections include Helicobacter pylori, hepatitis C virus, Malassezia, and Chlamydophila pneumoniae [5]. The connection between H. pylori and PD has long been discussed, in a way even before the discovery of H. pylori, as a link between peptic ulcers (of which H. pylori is a major causal agent) and PD was discovered in the 1960 s [61]. In more recent years H. pylori infection has been found to be more prevalent in PD populations and can affect PD motor symptoms, with the eradication of H. pylori improving PD motor dysfunction [62, 63]. Some have posited that H. pylori infections could be causal or at the very least a promoter of the development of PD [64]. PwP have been shown to also be more likely to be seropositive for several other bacteria including Borrelia burgdorferi and C. pneumoniae than healthy controls [65]. Furthermore, individuals who have previously been hospitalized for CNS infection [60] are at increased risk of PD as are those who have experienced GI infections caused by viral or bacterial pathogens [10].
While inflammations’ influence and perpetuation of PD processes is generally well accepted, some believe there is merit to inflammation as a disease initiator. Garretti and colleagues demonstrated that α-syn immunization of a specific HLA genetic mouse model leads to many GI features of PD, including constipation and enteric inflammation. These effects were mediated by CD4+ T cells and therefore the immune response promoted a PD-like phenotype [66]. Matheoud and colleagues described similar autoimmune processes that could be initiating neuronal dysfunction. They demonstrated in vivo that infection triggers antigen presentation of mitochondrial self-proteins. This presentation leads to the development of mitochondria-specific cytotoxic T cells in the periphery and brain, loss of synapses in the striatum, and motor dysfunction [47]. These studies demonstrate how inflammation need not just play a role in perpetuating the disease, but could initiate PD.
THE MICROBIOTA-GUT-BRAIN AXIS IN PD
The gut has long been of interest in PD research, as several lines of clinical and preclinical evidence converge upon a theory that the gut is a critical site of PD etiopathogenesis for some PwP [16]. In the prodromal stages of the disease (before the emergence of motor symptoms) GI symptoms, in particular mild constipation, are common [67, 68]. PD is associated with inflammatory bowel diseases (IBD) as they share common genetic risk factors, like LRRK2, CARD15, and ABCB1; and some groups of people with IBD are at a greater risk of developing PD [69, 70]. GI inflammation has been shown to accelerate the onset and exacerbate PD motor symptoms, influence α-syn aggregation, promote neuroinflammation, and exacerbate dopaminergic cell death in the midbrain in PD animal models [71–73].
Similarly to the pathological features of the CNS in PD, inflammation and α-syn inclusions occur also in the enteric nervous system [17, 74–83]. α-syn is proposed to be capable of transsynaptic spreading, trafficking via the vagus nerve to seed α-syn pathology in the CNS, as demonstrated in several animal model experiments [84–87]. These findings have led to some researchers proposing that PD could begin in the gut. The hypothesis revolves around the notion that initial pathology is triggered by an infectious agent or toxin, resulting in an inflammation-driven process that promotes α-syn misfolding within the gut and subsequent spreading of pathogenic α-syn from the gut to the brain via the vagus nerve [85, 89]. Other features of GI dysfunction are established including increased gut permeability (or “leaky gut”), increased bacterial invasion, and oxidative stress, which could be contributing factors and consequences of GI inflammation [83, 90]. While the gut is a critical site of etiopathogenesis in PD, whether disease begins in the gut is still a matter of contention.
An altered microbiome in PD
Since 2015, our understanding of the microbiota-gut-brain axis has evolved after several groups found that PwP exhibit an altered gut-microbiota compared to healthy controls [91–93]. Since then, at least 30 individual peer-reviewed studies have been published confirming these results [14, 94–126]. Recent meta-analyses taking into account many of these studies support PD gut dysbiosis as a robust finding, even when taking into account study design and geography [127, 128]. The most consistent findings in the PD gut microbiome are a lower abundance of protective anti-inflammatory bacteria genera, such as Roseburia, Blautia, and Faecalibacterium, and a greater relative abundance of potentially deleterious bacteria genera like Akkermansia. [14, 128]. Several of these studies have found significant correlations between elevated pro-inflammatory or opportunistic pathogen genera, such as Escherichia, Klebsiella, and Porphyromonas [124]. The cause of intestinal microbial dysbiosis in PD is not fully understood, but changes to the microbiota can be caused by aging, diet, heavy metals, toxins, and pesticides; which are also known environmental risk factors and triggers for PD [129–133].
An altered microbiome is not considered to be a simple consequence of the disease, but an active contributor to its progression [112]. Animal studies have demonstrated a role for the microbiota influencing gut permeability, local, systemic, and neural inflammation, enteric nervous system (ENS) and autonomic nervous system (ANS) signaling, microglial development, BBB integrity, and α-syn misfolding [134–137]. The reduction of anti-inflammatory bacteria involved in the maintenance of barrier integrity and immune regulation could lead to a greater susceptibility to pro-inflammatory insults, either from other harmful gut bacteria, pathogens, or environmental toxins [128]. Furthermore, specific changes to the gut microbiota have correlations with the severity of PD [98, 112].
Evidence of early changes in the microbiome further supports a role for microbial dysbiosis as a functional contributor to PD. Several studies investigating recently diagnosed and drug-naïve PwP have confirmed that microbiotic dysbiosis is not a late phenomenon and is not due to antiparkinsonian drugs [95, 121]. Most compellingly, studies that have explored the gut microbiota of people with rapid eye movement sleep behavior disorder (RBD), the most specific symptom predictor of future PD diagnosis, have shown that they also exhibit microbial dysbiosis that is more similar in composition to people diagnosed with PD that it is to healthy persons [101, 126]. This again supports that microbial dysbiosis is a feature of PD that manifests early.
Aside from changes to the gut microbiota as measured from fecal samples, small intestine bacterial overgrowth (SIBO) is another form of dysbiosis that is commonly diagnosed in PD [138]. SIBO is known to impact on the absorption of levodopa, subsequently impacting on motor function, and is associated with greater disease severity [139, 140]. SIBO can promote GI inflammation and weaken barrier integrity, which can expose the local and peripheral immune system to harmful bacterial and non-bacterial agents [141, 142]. The connections between PD and SIBO highlight another way in which bacteria may be relevant to facilitating disease processes.
in vivo studies have demonstrated the importance of the gut flora to the development of disease, showing that PD mouse models that would normally develop motor deficits, α-syn aggregation, and microglial activation do not develop these phenotypes when raised in a germ-free environment [135]. The same group also showed that germ-free PD model mice have restoration of the PD phenotype after colonization of their GI tract with a microbiota derived from PwP or healthy controls, however, the severity of the PD phenotype was greater after the colonization with PwP-derived microbes [135].
Inflammation and the microbiota-gut-brain axis in PD
Although it is difficult to dispute that the microbiota plays a role in PD, the question remains: how does a shift in intestinal microbial diversity facilitate the progression of disease? In good gut health, most gut bacteria act indirectly on host cells as they remain in the gut lumen, separated from the epithelium by a thick mucosal layer that protects against their invasion [143, 144]. Being predominantly restricted to the gut lumen, in order to communicate with host cells, such as epithelial and immune cells, bacteria must act indirectly via the release of secreted factors including OMVs.
Bacterial metabolites and secreted factors such as short-chain fatty acids (SCFAs) and toxins (such as lipopolysaccharide (LPS)) have been investigated as mechanisms by which microbiota affect global changes in disease, in particular in PD [94, 145–147]. LPS, the major component of the outer membrane of Gram-negative bacteria and a potent activator of the innate immune response, has been shown to be increased in the blood of PwP [148, 149]. This finding is substantiated by research indicating higher levels of the LPS binding protein (LBP) in serum during the prodromal phase of the disease [150]. This connection extends beyond systematic effects, as demonstrated by increased expression of the microbial-associated molecular pattern (MAMPs) receptors, specifically toll-like receptor 4 (TLR4) and toll-like receptor 2 (TLR2), in postmortem PD brain tissue [151–153]. For many years LPS has been used to model neurodegenerative disease [147]. PD researchers use LPS (typically from E. coli strains) to elicit a neuroinflammatory response that ultimately leads to neurodegeneration of the dopaminergic neurons of the SN [154–156]. This response is mediated by the activation of microglia and the subsequent release of pro-inflammatory cytokines and destructive reactive oxygen species [32, 157]. LPS injections directly into the SN (and not other sites in the brain) elicit this response, indicating that LPS can direct PD-specific neurodegenerative processes [155, 156]. LPS administration has been shown to increase α-syn expression in wild-type animal ENS and SN, exacerbate α-syn aggregation in PD models, and make animals more susceptible to neurotoxins [158–161]. When administered systemically at a low dose, LPS models the neuronal features of PD, as well as GI features such as increased intestinal permeability and increased α-syn expression in the colon [160].
LPS found on E. coli strains and other Enterobacteriaceae members, are considered to be the most immunogenic forms of LPS [162, 163]. Notably, Enterobacteriaceae have an increased presence in the gut microbiota of PwP along with other Gram-negative LPS-producing families Veruccomicrobioaceae and Christensenellaceae [93, 123] and Klebsiella species, and Porphyromonas asaccharolytica [124]. In the field of neurodegenerative disease, there has been a prevailing assumption that secreted, non-vesicle-associated factors, such as free LPS, breach the intestinal barrier and trigger an immune response that contributes to disease progression. We propose an expansion of this hypothesis to include a crucial role for OMVs in this process. Bacterial OMVs have yet to be investigated in the context of PD despite being a major secretion and communication pathway for bacteria, being highly immunogenic, and able to deliver cargo across biological barriers like the gut and BBB [164, 165]. For visual hypothesis see Fig. 1.
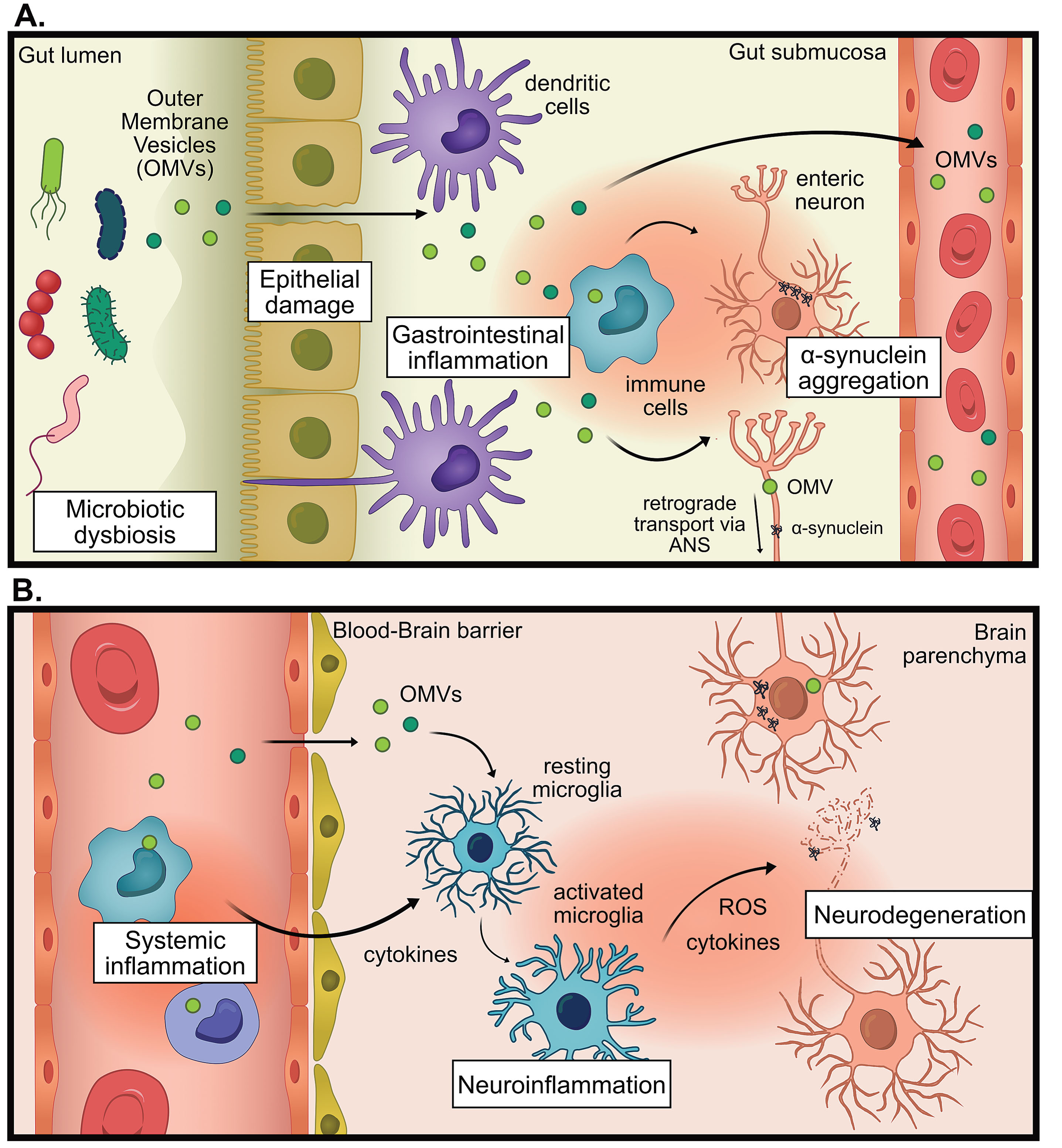
The proposed hypothesis for the pro-inflammatory role of outer membrane vesicles (OMVs) in Parkinson’s disease (PD). A) In states of microbial dysbiosis, OMVs and other microbial factors can cause damage to epithelial barriers, causing the translocation of OMVs past the epithelium, where immune cells can be activated to promote gastrointestinal inflammation which increases intestinal permeability. Information about gastrointestinal inflammation can be directly communicated to the brain via the autonomic nervous system (ANS), by altering neural signaling or retrograde transport of pathogenic proteins (for example α-synuclein) or OMVs. B) OMVs reach the systemic circulation whereby they promote systemic inflammation. OMVs could promote neuroinflammation indirectly via increasing systemic inflammation, leading to weakening of the blood-brain barrier, entry of immune cells into the brain and increased cytokine signaling. OMVs could also promote neuroinflammation directly by infiltrating the brain and activating microglia. This chronic inflammation can contribute to neurodegeneration.
BACTERIAL OUTER MEMBRANE VESICLES
OMVs are nanosized membranous structures that range from 20–200 nm and have known roles in bacterial communication and survival [166]. These vesicles are secreted as distinct entities to mediate intercellular communication between bacteria and their host cell tissues [167]. OMVs are complex structures that are comprised of many different antigenic bacterial proteins, lipids, and nucleic acids on their membrane surfaces and also as cargo within their lumen. The extracellular vesicles released from Gram-negative and Gram-positive bacteria are generally termed OMVs and membrane vesicles (MVs), respectively; however, nomenclature variation exists in the literature [166]. For the purpose of this review, we will focus on the extracellular vesicles released from Gram-negative bacteria, which we will refer to as OMVs; however, we do acknowledge the potential relevance of Gram-positive MVs in disease also (for a review, see [168]).
There are multiple mechanisms by which Gram-negative bacteria shed OMVs, such as envelope crosslink modulation, envelope component accumulation, and insertion of particular lipids into the outer membrane, the importance of this being that OMV release is not a stochastic event, and highly regulated by the cell (reviewed in [169]). Depending on their bacterial source, OMVs have varied functions that are related to their composition, for example, delivery of toxins, horizontal gene transfer, and biofilm formation (see review by Gilmore et al. for further information [170]). Unlike the parental bacterium, OMVs are not so restricted to the gut lumen and can traverse tight junctions to deliver their payload over long distances within the host, while protecting vesicular contents from the external environment [171, 172]. Bacterial vesicles have been detected in a multitude of tissues and fluids including blood and plasma [173–175], cerebrospinal fluid [176, 177], gastric mucosa [178], urine [179–181], saliva [179] and feces [174, 182–184], indicating their widespread biodistribution, including in the brain [185, 186].
The role of OMVs in immune modulation
One major function attributed to gut-derived OMVs that is relevant to PD is host immune system modulation. OMVs, released by bacteria in the gut, can trigger local and distant proinflammatory immune activation by interaction with both innate and adaptive immune cell types including, macrophages, dendritic cells, neutrophils, and B cells [187–194]. Enclosed by a membrane, OMVs harbor many types of inflammogens such as bacterial DNAs, RNAs, peptidoglycan, lipoproteins, LPSs, and toxins, that can result in a complex multimodal mechanism of immune activation [195]. Porphyromonus gingivalis and Aggregatibacter actinomycetemcomitans OMVs have been demonstrated to interact with neutrophils by coating their plasma membrane, and are also internalized by macrophages and epithelial cells [190]. Once bound to neutrophils P. gingivalis OMVs induce cell activation but also degrade neutrophil effector protein myeloperoxidase, which results in greater survival of the P. gingivalis parent bacterium [190]. OMVs from E. coli as well as total bacterial extracellular vesicles from feces have been demonstrated to cause sepsis-like inflammation in rodents after intraperitoneal injection or intravenous infusion [183, 196–198]. In high enough doses, the sepsis-like effects caused by OMVs can be lethal, in contrast, there is no lethality when the animals are administered a dose of pure LPS that is more than double the LPS dose contained in the OMV preparation [197].
While OMVs contain many MAMPs, a major component is their LPS-rich outer membrane [164, 199]. OMVs, studded with LPS on their surface are able to deliver LPS to many types of cells [200], including microglia [28]. LPS predominantly activates immune cells by binding extracellularly to TLR4/CD14/MD2 receptor complex, which results in intracellular NF-κB signaling and thus the release of pro-inflammatory cytokines, TNF-α, IL-1β, and free radicals, nitric oxide and superoxide [201]. However, OMVs have been identified as a method of delivering LPS intracellularly where they activate caspases and the NLRP3 inflammasome [200]. Several E. coli strains (enterotoxigenic and avirulent strains) release OMVs that can be genotoxic, contain a heat-labile toxin, and can confer protection to the producer bacteria and others against some antibiotics [195, 203]. E. coli OMVs have been shown to induce mitochondrial dysfunction, mitochondrial apoptosis, and activate inflammation, which is particularly relevant in PD, as mitochondrial impairment is characteristic of the disease [204].
The finding of elevated pro-inflammatory Gram-negative bacteria and elevated synthesis of LPS in PwP may indicate that the gut microbiome in PwP produces a greater amount of OMVs which contain highly potent LPS. LPS has been shown to be increased in the blood of PwP [148, 149], however the methods employed in these studies did not differentiate free monomeric LPS from LPS contained on OMVs, suggesting LPS found in the bloodstream of PwP could be OMV-associated.
OMVs interact with cells either by ligand binding to externally expressed pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) or intracellular receptors like nucleotide-binding-oligomerization domain-containing proteins (NOD) [188, 205–207]. In the context of PD, the major PRR families that are involved in sensing MAMPs, (TLRs, and Nod-like receptors (NLR)), are implicated in the disease [208]. Leukocyte TLR2 responses are lowered in PD compared to controls. This could indicate a reduced ability to respond to bacterial lipoproteins and other TLR2 ligands [209]. Conversely, leukocyte TLR4 responses are increased in young PwP. When stimulated with LPS, leukocytes from these individuals have a more robust TNFα cytokine release than healthy controls, indicating that in PD there is an overactive proinflammatory response early in the disease [209]. The expression of TLRs is increased in the brain, (particularly in the caudate putamen), blood, and GI of PwP [83, 153]. One study found that TLR2 expression is found to be increased in the neurons of PwP, and in particular, neurons harboring aggregated α-syn [151]. Another study demonstrated that the SN, a brain region highly susceptible to neuroinflammation, is one of the regions with the highest TLR4 expression [152]. NLR family proteins, such as NOD2 which detects peptidoglycan, and the NLRP3 inflammasome complex which detects a wide variety of MAMPs (as well as damage-associated molecular patterns) have also been implicated in PD [39, 211]. Levels of NLRP3 are increased in the serum and peripheral blood mononuclear cells of PwP and these levels positively correlate with the level of serum α-syn [39, 210]. Variant alleles in the gene that encodes NOD2 (CARD15) are associated with an increased risk of PD [211]. Both NLRP3 and NOD2 detect OMVs and their components, for example, NLRP3 detects E. coli OMV-delivered LPS and NOD2 detects OMVs from A. actinomycetemcomitans [200, 205].
The changes to TLR expression and other PRRs could indicate that PwP may be differentially able to detect bacterial components, like OMVs, and that they may produce aberrant immune responses which could contribute to disease as a result. TLRs are mostly expressed in immune cells, including microglia, but neurons, in both the gut epithelium and brain, also express TLRs [212–214]. This naturally leads to the implication that OMVs may be capable of directly interacting with neurons (should they come in contact), however, this has not yet been investigated to the best of our knowledge.
OMVs and the epithelium
OMVs released in the gut lumen can pass through the thick mucosal layer and associate with epithelial cells, where they can initiate cytokine release, affect cell growth, and affect tight junction protein expression [178]. OMVs are known to pass through the epithelium either paracellularly or transcellularly to access the lamina propria, where they can interact with local immune cells [164, 215]. OMVs from bacteria such as Desulfovibrio fairfieldensis, Campylobacter jejuni, H. pylori, and P. gingivalis have been found to reduce the expression of important tight junction proteins, like occludin, zonulin-1, and E-cadherin, which results in the weakening of the epithelial barrier and could potentially promote their own translocation from the lumen into deeper layers of the gut and beyond [175, 216–219]. Tulkens and colleagues demonstrated that there were increased bacterial extracellular vesicles in the plasma of people with intestinal barrier dysfunction compared to healthy controls and that the bacterial extracellular vesicle concentration was associated with greater plasma zonulin-1 levels (indicating intestinal barrier dysfunction). They also demonstrated that in an in vitro colitis model that bacterial extracellular vesicle concentration was increased in the basal side as zonulin-1 levels decreased, indicating paracellular translocation from the apical side where they were applied [175]. This role in affecting epithelial permeability is particularly relevant in PD as elevated intestinal permeability and the translocation of bacterial products occur in PwP.
OMVs beyond the gut
It is unlikely that major OMV translocation beyond the gut lumen is an innocuous event as OMV’s ability to influence the function of many cell types could have flow-on effects. The ability of OMVs to travel throughout the body allows for bacteria restricted to the gut to elicit long-range effects indirectly, permitting them to functionally interact with the important sites of PD pathology, mainly the gut and potentially the brain. A study investigating OMV biodistribution demonstrated that labeled OMVs administered orally to mice are predominantly found in the GI tract and liver and OMVs delivered intraperitoneally are predominantly found in the liver, and less so in the spleen, kidneys, and lungs (this study did not investigate the brain) [171]. This study was performed in healthy mice and therefore supports that OMVs can pass through the gut even without gut impairments [171].
As described above OMVs are highly effective immune activators and their presence in the circulation and distant organs has major implications for the facilitation of inflammation systemically. We acknowledge the possibility that OMV translocation from the gut lumen may not occur in sufficient quantities to affect systemic and neural inflammation directly, however, it is plausible that infiltration of OMVs into the lamina propria and submucosa may promote local inflammation and alterations to ENS signaling to an extent that results in a modulation of the peripheral immune system and CNS signaling. OMVs, in this case, need not traverse into the vasculature or brain, while still contributing to PD pathogenesis.
The presence of OMVs in the bloodstream and urine has been established in other conditions and biodistribution studies and this supports that OMVs can pass through epithelial layers to traverse into the vasculature [171, 220]. It has been demonstrated that individuals with GI barrier disruption have a greater number of bacterial extracellular vesicles in their circulation compared to healthy controls [175]. Though this has yet to be tested in PwP, it stands to reason that bacterial extracellular vesicles could similarly be elevated in the circulation, because of the evidence of increased intestinal permeability or reduced gut tight-junction proteins in PwP [90, 222].
OMVs and the brain
Several research groups have hypothesized that OMVs can cross the BBB, especially in cases where the BBB is impaired, theorizing based on the nanosize of OMVs and citing evidence of bacterial components such as bacterial nucleic acids and LPS being found in brains, particularly of those with neurological dysfunction [165, 223]. It has been demonstrated that OMVs and LPSs, from species like E. coli and P. gingivalis, can impair endothelial barriers including the BBB, which would perhaps promote uptake of OMVs or other toxic materials, allow bacterial infiltration into the brain and also promote neuroinflammation via peripheral immune cell infiltration [224–230]. The BBB in PD has been suggested to be dysfunctional, as evidenced by positron electron tomography hyperpermeability experiments, investigations of cerebrospinal fluid-serum albumin ratios, and also histological investigation of BBB integrity markers [231–233]. While the cause of the BBB dysfunction is unknown in PD, these observations imply that the BBB may be susceptible to OMV penetration.
There is the potential that OMVs could cross an intact BBB, as eukaryotic EVs have been suggested to do [234], or in a similar manner to how they transcellularly cross epithelial and endothelial cells [171]. Evidence for OMVs crossing the BBB in animal models is beginning to emerge [185, 235–237]. A. actinomycetemcomitans OMVs can cross the BBB, deliver bacterial RNA, and induce the release of cytokines following intracardial administration [235]. In a follow-up study, OMVs were shown to be taken up by microglia and meningeal macrophages [186]. Orally-gavaged OMVs from Paenalcaligenes hominis are delivered to the hippocampus via the autonomic nervous system resulting in cognitive impairment more potently than orally-gavaged LPS [238]. To the best of our knowledge, this study is the first to experimentally link OMVs and neuronal dysfunction in the CNS, showing retrograde delivery from the gut to the brain via the vagus nerve, which is believed to be an important conduit between the gut and the brain in PD and links the gut to key sites of PD pathology, such as the dorsal motor nucleus [238–240]. It also highlights the possibility of a route for gut-to-brain trafficking of OMVs that bypasses the circulation and thus avoids the challenges of evading the peripheral immune system and crossing the BBB.
In another study, which provided evidence of OMV passage into the brain, H. pylori-derived OMVs were shown to pass into the brain through the transcellular method [185]. This study also demonstrated that by orally delivering OMVs to Alzheimer’s model mice, the OMVs are taken up by astrocytes. Notably, OMVs increased plaque load, inflammation, neuronal dysfunction, and also accelerated cognitive decline [185]. H. pylori-derived OMVs have also been demonstrated to traffic to the mouse brain following venous and oral administration, where they activate astrocytes and cause neuronal damage [237]. As previously discussed, there is are connection between H. pylori infection and PD, therefore an investigation into H. pylori-specific OMVs in PD has sufficient merit. A separate study investigating Alzheimer’s disease demonstrated that after oral gavage of P. gingivalis OMVs into middle-aged wild-type mice, the OMVs reached the brain, impaired expression of tight junction proteins, were able to induce inflammation, and impaired learning and memory [241].
While the studies described above use non-physiological quantities of OMVs in acute animal models, throughout the lifetime of a person, minute insults from gut-derived OMVs could contribute to PD by fueling a state of chronic inflammation, ultimately contributing to neurodegeneration over the course of decades.
CONCLUDING REMARKS
The etiology and pathogenesis of PD are multifactorial, and the interaction between age, genetic susceptibility, environmental influence, and microbial function all appear to contribute to PD pathogenesis. There is increasing appreciation of the importance of the microbiome in health and disease. A greater understanding of bidirectional gut-brain axis functions will allow for more sophisticated examinations of the interaction between gut and brain health in systemic diseases such as PD.
A multitude of studies demonstrate that models using inflammogens, like LPS, recapitulate the neuroinflammation and neurodegeneration seen in PwP. There is an abundance of evidence supporting changes in the GI tract of PwP including increased intestinal permeability, microbial dysbiosis, and evidence of increased LPS in blood. Thus, a hypothesis has formed that people with GI dysfunction have greater amounts of LPS reaching their circulation, which promotes systemic inflammation and promotes neurodegeneration. OMVs are a physiological mechanism by which MAMPs are released from bacteria to promote immune responses. Considering their presence in the bloodstream and potential ability to cross the BBB, we propose a more likely hypothesis, that OMVs released from bacteria in the gut trigger inflammation to contribute to the progression of PD.
The implication of this hypothesis opens new avenues for therapeutic interventions in PD and potentially other diseases where OMV-induced inflammation plays a role. This is because OMVs elicit distinct host immune responses so therapeutic approaches specifically tailored for soluble LPS for example, may prove inadequate in addressing inflammation triggered by OMVs [242]. Re-establishing a healthy microbiome and strengthening gut barrier integrity could be one mechanism for reducing OMV-induced inflammation. TLR4 and other OMV-detecting PRR antagonists could prevent increases in inflammatory processes. Lastly, OMVs could provide a new avenue for biomarker development with the difference in the relative abundance of plasma OMVs used as a complementary diagnostic tool for PD.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The Florey Institute of Neuroscience and Mental Health acknowledges the support from the Victorian Government and in particular funding from the Operational Infrastructure Support Grant. We acknowledge an Australian Government Research Training Program Scholarship (TK).
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.