
Abstract
Regulation of gene expression by epigenetic modifications means lasting and heritable changes in the function of genes without alterations in the DNA sequence. Of all epigenetic mechanisms identified thus far, DNA methylation has been of particular interest in both aging and age-related disease research over the last decade given the consistency of site-specific DNA methylation changes during aging that can predict future health and lifespan. An increasing line of evidence has implied the dynamic nature of DNA (de)methylation events that occur throughout the lifespan has a role in the pathophysiology of aging and age-associated neurodegenerative conditions, including Parkinson’s disease (PD). In this regard, PD methylome shows, to some extent, similar genome-wide changes observed in the methylome of healthy individuals of matching age. In this review, we start by providing a brief overview of studies outlining global patterns of DNA methylation, then its mechanisms and regulation, within the context of aging and PD. Considering diverging lines of evidence from different experimental and animal models of neurodegeneration and how they combine to shape our current understanding of tissue-specific changes in DNA methylome in health and disease, we report a high-level comparison of the genomic methylation landscapes of brain, with an emphasis on dopaminergic neurons in PD and in natural aging. We believe this will be particularly useful for systematically dissecting overlapping genome-wide alterations in DNA methylation during PD and healthy aging, and for improving our knowledge of PD-specific changes in methylation patterns independent of aging process.
KEY CONCEPTS IN PARKINSON’S DISEASE
Pathologically described as selective loss of dopamine (DA) neurons of the substantia nigra pars compacta in the midbrain and formation of α-synuclein protein aggregates referred to as Lewy bodies, Parkinson’s disease (PD) is among the most common nervous system disorders of the elderly, most often diagnosed in people over age 60 [1]. The clinical manifestations of PD include rigidity, tremors, slowness/absence of voluntary movement, and posture instability, as well as intellectual deficiency, mental breakdown, sleep, and mood disorders [2]. Although great effort has been devoted to understand the genetic determinants of PD, the findings could explain only a fraction of the cases. As in other age-related neurodegenerative disorders, the existence of an epigenetic component contributing to the disease phenotype has been largely considered and extensively studied in PD, as well [3, 4]. In this regard, genome-wide changes in DNA methylome observed in both PD and healthy aging of similar age bear some resemblance, such as global hypomethylation with promoter-specific patterns of differential methylation [5–9]. This review discusses global patterns of DNA methylation, its mechanisms and regulation, within the context of aging and PD, and define the genomic methylation landscapes of brain, with an emphasis on dopaminergic neurons, in PD and in natural aging to dissect the extent to which DNA methylation changes in the SN with these two interrelated events of human senility.
FACTORS INFLUENCING THE AGING PROCESS
Aging refers to a time-dependent decline of both biological functions and of the organism’s ability to handle metabolic stress leading to both physical deterioration and increased vulnerability to death. Accumulation of somatic mutations, the resulting genomic instability, ongoing telomere shortening, compromised protein homeostasis, discordance of mitochondrial network, impaired nutrient sensing, decline of regenerative capacity, and disorganized cell-to-cell signaling including inflammation are among the preeminent factors that contribute to the aging process [10]. In this context, mitochondrial dysfunction has long been of interest in biomedical research due especially to its causative implications for neurodegenerative disorders [11–13]. Aside from these deteriorations at the molecular, cellular, and physiological levels with age, epigenetic alterations that act potentially on different cell types with varying degrees of magnitudes throughout the lifespan is a hallmark of aging [14]. The progressive loss of original epigenetic configurations observed during aging, also called “epigenetic drift”, is an active field of research [15].
DNA METHYLATION
A historical perspective
An organism’s ability to regulate and maintain the patterns of tissue-specific gene expression temporally and spatially in response to changing environmental cues reflects how well an organism is adapted to its surrounding [16]. To fine-tune this ability in a reversible and heritable manner without mutating the genetic sequence provides not only a unique flexibility in modulating transcriptome at any given instance but also a new route of abiding marks of former exposures which are acquired throughout the lifespan [5]. The idea of the epigenetic landscape was initially proposed as an “abstract strategy” of the genes to regulate embryonic development more than 60 years ago [17]. Afterwards, the regulatory role of epigenetics has been increasingly observed at later stages of life, as well [18]. In that sense, recent advances in epigenetics have had enormous impact on our understanding of the contribution of environmental stressors to complex phenotypes [19], thereby putting epigenetics under spotlight in the context of the etiology of age-related disorders, notably waning and neurodegeneration. It is now widely accepted that epigenetic mechanisms provide an additional layer of transcriptome regulation that combines environmental factors with the genetic elements to achieve a concerted functional outcome toward better fitness or beneficial phenotypes [20]. DNA and chromatin modifications, as well as non-coding RNAs, constitute the cardinal epigenetic regulatory mechanisms in mammalian cells [6]. This review focuses particularly on alterations in DNA modifications considering the association of the dynamic nature of DNA (de)methylation with the pathophysiology of aging and age-associated neurodegenerative conditions, especially PD [6, 8]. Also, DNA methylation has become a central theme of both aging and age-related disease research after many groups independently identified a highly significant association of site specific changes in CpG methylation with aging in different tissues [21–26], with implications for predicting future health and lifespan [5, 27]. These findings led to the discovery of the “epigenetic clock”, which is another area of active research [15].
Basics of DNA methylation
The most highly studied phenomenon in epigenetic modifications by far is DNA methylation [28], which typically refers to covalently attaching a methyl group (CH3) to the 5th position of the cytosine nucleotide by means of a group of specific enzymes called DNA methyltransferases (DNMTs) using S-adenosyl-L-methionine (SAM) as substrate [29, 30]. Though several studies identified the non-CpG methylation across many different tissues including brain, cytosine methylation in the mammalian genome is almost exclusively restricted to the symmetrical CpG (5′— C— phosphate— G— 3′) dinucleotide [31, 32]. In this sense, methylated cytosine is regarded as the 5th base of DNA, expanding the genetic alphabet into the realm of epigenetic modifiers [33]. In humans, methylation is observed sparsely but globally, either interspersed as single/few copies (∼90% of all) of CpG dinucleotides throughout the genome or concentrated into “CpG islands” (CGIs), which are approximately 1kb CpG stretches in vicinity of promoter regions [34]. CGIs mostly lack DNA methylation but the flanking regions to the CGIs, called CGI shores (0–2 kb from island edge) and shelves (>2–4 kb from island edge), are vulnerable to tissue-specific methylation, which was found to correlate with alterations in mRNA levels [35–39]. Overall, of a total of around 28 million CpGs in the human genome [40], 60–80% are found methylated at any given time, unlike those in CGIs [41].
DNMT3a-b together with DNMT3l function to establish de novo patterns of DNA methylation during differentiation [29, 42], while DNMT1 functions as maintenance methyltransferase detecting hemi-methylated DNA following genome replication during mitosis and meiosis [30, 43]. In this sense, SAM, which acts as a methyl donor at this point, is the substrate of a critical reaction in DNA methylation (Fig. 1A). The discovery that the Ten-eleven translocation family of protein dioxygenases (TET1-3) can convert methylated cytosines (5-methylcytosine, or simply 5mC) into 5-hydroxymethylcytosine (5hmC; also called the 6th base of DNA due to its high abundance in the genome), as well as 5-formylcitosine (5fC) and 5-carboxylcytosine (5caC), in a process called DNA demethylation has implicated dynamic regulations of DNA modifications is a natural paradigm in which to account for intermediary epigenetic states [44, 45]. Historically, DNA methylation was considered as an irreversible modification used as a one-way, static tool rather than a bidirectional, dynamic modification of DNA [46]. It was in 2009 that the intermediary states of methylated cytosine discussed above as the products of TET-catalyzed reactions were identified [45, 47], enabling the possibility of restoring back to the unmethylated state of the cytosine [44, 48]. Together, DNMTs and TETs work concertedly to regulate the dynamics of DNA methylation in mammalian cells.
Carousel Figure: Dynamic DNA methylation in aging and Parkinson’s Disease.
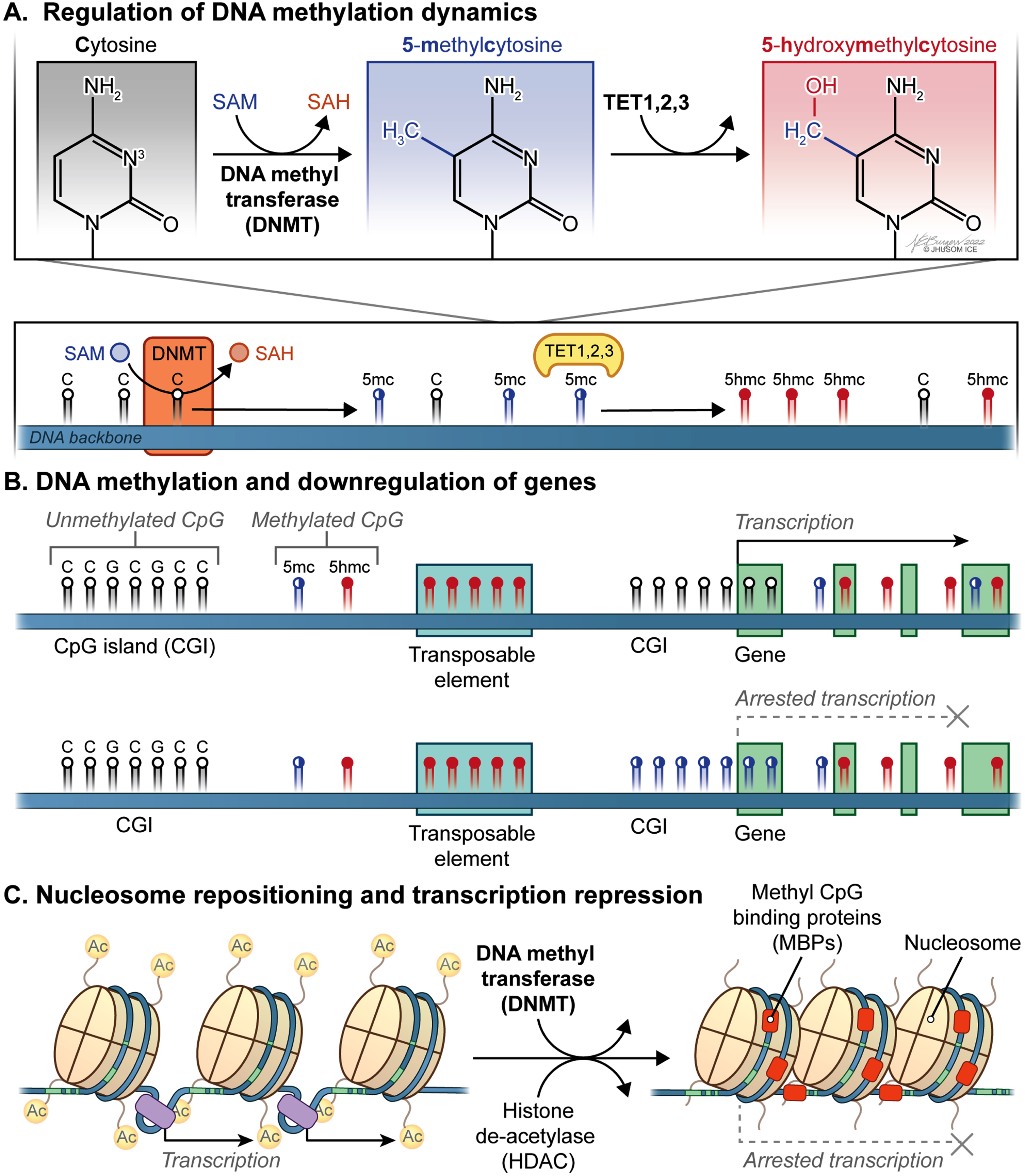
Dynamic CpG methylation landscape of the mammalian genome within the context of transcriptional regulation. A) S-adenosyl-L-methionine (SAM) acts as a methyl donor for a critical reaction in DNA methylation catalyzed by DNA methyltransferases (DNMTs), which can be reversed by Ten-eleven translocation family of protein dioxygenases (TETs). B) Genomic methylation involving promoter and/or distal regulatory regions plays a key role in downregulation of the corresponding genes. Also, epigenetic silencing of transposons and other genomic elements that undermine genomic integrity occurs in mammalian cells. C) Nucleosome repositioning and transcription repression involve the concerted actions of DNMTs and histone de-acetylases (HDACs).
Reprogramming of mammalian transcriptome by DNA methylation
As previously discussed [49], many fundamental physiological processes, including X chromosome inactivation in the early female embryo [50, 51], genetic imprinting [52, 53], epigenetic silencing of transposons and other genomic elements that undermine genomic integrity [54, 55], and preservation of constitutively repressed structural DNA repeats [56] are achieved by DNA methylation. Perhaps most critically, genomic methylation involving promoter and/or distal regulatory regions plays a paramount role in downregulation of the corresponding genes considering how vital cis-acting DNA is for proper transcriptional activity [33] (Fig. 1B). More specifically, a distinct set of mechanisms underlying transcriptional repression by DNA methylation has been identified, which includes alterations in the affinity with which target DNA is bound by transcription factors (TFs), remodeling and repositioning of nucleosomes (Fig. 1C), and the physical interactions between methyl CpG binding proteins (MBPs) and the genomic DNA [49]. For instance, some of the common TFs, such as CCCTC-binding factor (CTCF) [57], CREB [58], c-Myc [59], E2F1 [60], and Usf1 [61], show significantly reduced binding affinity to the corresponding target sequences after methylation at the cytosine residues. Currently, the impact of DNA methylation on nucleosome dynamics, which is an indirect way of transcriptional regulation [62], appears not fully settled. Practically, this is a reciprocal relationship discussed elsewhere [63]. As a final way of DNA methylation-based transcriptional repression, CpG methylation paves the way for MBPs to assemble repressive chromatin modifiers for transcriptional downregulation locally in mammalian cells [64]. On the other hand, it has been repeatedly shown that a methylated gene body suggests increased levels of expression [65–67]. Together, methylation of CpGs across the genome is a pivotal regulatory mechanism contributing to transcriptome reprogramming in response to internal or external cues in a timely and flexible manner.
Changes in the DNA methylome of PD brain
Considering the growing body of evidence that there is an epigenetic component in the pathogenesis of PD, research on likely disruptions in DNA methylation dynamics of DA neurons can improve our understanding of the SN-specific neuronal death observed in both sporadic and familial PD. Contrarily, this rationale could not be expanded to cover all aspects of the discordant methylation patterns identified in PD yet. Additionally, as in the literature on age-dependent DNA methylome changes in brain below, most of the current research investigating promoter-specific alterations in CpG methylation in the SN of patients with PD is far from reaching a consensus due to lack of clarity. Generically, a group of methylation anomalies in CpG sites have been linked to aberrant gene silencing or reactivation, including downregulation of neuroprotective genes and overexpression of neurotoxic genes, in PD (Fig. 3). For instance, the α-synuclein (α-syn, SNCA) promoter is hypomethylated in PD [26], which contributes greatly to the phenotype by promoting overexpression of this gene as a neuropathological hallmark of the disease [68, 69]. From a mechanistic perspective, misfolded α-syn has been demonstrated to initiate retention of DNMT1 in the cytoplasm of neuronal cells in PD [70]. This observation might account for the molecular underpinnings of the global change in DNA methylome in favor of hypomethylation in PD. Still being the primary pharmacological treatment in patients with PD 40 years after its first use [71], L-dopa treatment has been associated with restoration of the methylation status of SNCA promoter back to its hypermethylated state, implying traditional PD therapy acts on DNA methylome in human neurons [72].
Though being the quintessential example of how aberrant DNA methylation contributes to PD-associated neurotoxicity, SNCA is not the only differentially methylated gene identified in this context. A hypermethylated state was identified in the SN of patients with PD for the promoter of PGC1-α, the master regulator of mitochondrial biogenesis and a key player in neuroprotection [73]. Mitochondrial dysfunction causatively linked to PGC-1α silencing along the NRF1/2-TFAM axis is an active field of research in PD [12, 13]. In this regard, CpG methylation has also been suggested to contribute more to age-related mitochondrial impairment by inducing production of reactive oxygen species (ROS) after transcriptional repression of the two PD-related genes, Parkin (PARK2) and PINK1 [74–76], though with limited empirical evidence. Other abnormal DNA methylation changes that lead to dysregulation of PD genes have also been reported [75, 77], including but not limited to hypomethylation of NPAS2 [78], CYP2E1 [79], VTRNA2-1 [80], NOS2 [81], and TNF-α [82], hypermethylation of H1 haplotype of Tau (MAPT) [83], GFPT1, and HLA-DQA1 [84]. From a biochemical standpoint, dynamic regulation of transcription by genomic methylation requires a steady supply of methyl groups. The universal substrate for DNMTs, SAM, is a methyl donor involved in the metabolism of methionine to homocysteine. Both methionine and folate cycles have been associated with parkinsonism after higher concentrations of homocysteine (a byproduct of the methyltransferase reaction) were detected in PD patients compared to controls [85]. For excellent reviews of metabolic factors related to DNA methylation and their implications for PD, please see [28, 75]. Overall, the most widely accepted mechanistic explanation for global hypomethylation associated with PD to date is the retention of DNMT1 in the cytoplasm in both post-mortem brain tissue of PD patients and SNCA transgenic mice.
Contrary to the studies cited above, several independent lines of research have reported conflicting results at the gene level. One study reported both hypo- and hypermethylation states at the SNCA promoter and intron 1 regions in various brain sections of patients at distinct stages of PD [86]. The research by [87] in 2017 identified no significant alterations in CpG methylation associated with SNCA intron 1 in PD patients. Samples from PD patients with or without heterozygous PARK2 mutations were used for a comparison with the matching samples from healthy individuals in a different study only to find no differential methylation at CpG sites, implying DNA methylation has no role in the etiology of a Parkin-induced PD phenotype [88]. Yet another study reported no differential methylation at the PARK2 promoter in post-mortem brain samples obtained from the SN, cerebellum, and occipital cortex of PD patients [89]. Without a consensus on likely association of CpG methylation with transcriptional regulation of several Parkinson’s linked genes, the hunt for novel loci associated with PD and the corresponding methylation levels is an active area of research [28]. The incongruity of findings on brain-specific CpG methylation signatures at several PD-linked genes is disconcerting given the clinical significance and the association of the dynamic nature of genomic (de)methylation with the pathophysiology of Parkinson’s disease. Most critically, the empirical proof of concordant changes in matching PD-related gene expression and genomic methylation in PD brain or in animal models is yet to be found. Therefore, the extent to which DNA methylation changes with PD in the SN and whether or to what degree PD-associated epigenetic dysregulation correlates with the transcriptome changes in the SN leading to dopaminergic degeneration remain to be investigated in future studies.
Navigating the DNA methylation landscape of aging brain
Epigenetic processes are central to complex brain functions. A wide range of physiological and cognitive activities in the cerebrospinal axis, including but not limited to development of nerve tissues, formation of functional neural networks, synaptic activities, brain plasticity, and learning, are all modulated epigenetically in mammals [90]. After the findings that one of the most highly methylated genomic DNAs was obtained from human brain (with 0.98-moles % 5-methylcytosine) and that brain is the second most methylated human tissue after thymus [91], DNA modifications have become one of the well-defined epigenetic processes in neuroscience over the past few decades [92–94]. In this respect, aging has mostly, if not always been a part of the equation, though with conflicting reports [6, 8]. The de novo DNA methyltransferase called DNMT3a has a continuous pattern of expression in human brain throughout the lifespan [66], enabling a potent epigenetic regulation of neural transcriptome at any stage of life [95]. The same enzyme was reported to have an increased level of expression in the mouse hippocampus, leading to genome-wide hypermethylation with age [96–99]. Age-associated global hypermethylation in murine cerebellum and cerebral cortex was also reported [100, 101]. There are other studies reporting similar findings [102].
On the other hand, a great majority of the studies support the view that mammalian tissues, including brain cells, show a global pattern of DNA hypomethylation (Fig. 2A) with many promoters undergoing aberrant hypermethylation with age [5, 103–109] (Fig. 2B). Activities of maintenance and de novo DNMTs, decreasing and increasing respectively, were proposed to explain this trend, at least to some extent. Indeed, there is a growing body of evidence that an epigenetic drift that alters the 5mC distribution in the genome is observed during healthy aging. These stochastically occurring epigenetic changes take place mostly in intergenic nonisland CpGs that overlap transposable regions, such as Alu and LINE-1 [8]. This in turn relaxes transposition activity, resulting in genomic instability [110]. The direct effect of this epigenetic drift is on the coding DNA: a general downregulation of gene expression with age leads to intracellular miscommunication and deregulation of vital biological processes [111]. Yet, it was also suggested that a key mechanism for transcriptome diversity and regulation of gene expression in brain, the alternative promoter usage, is independent of promoter methylation in different regions of the aged brain [112].
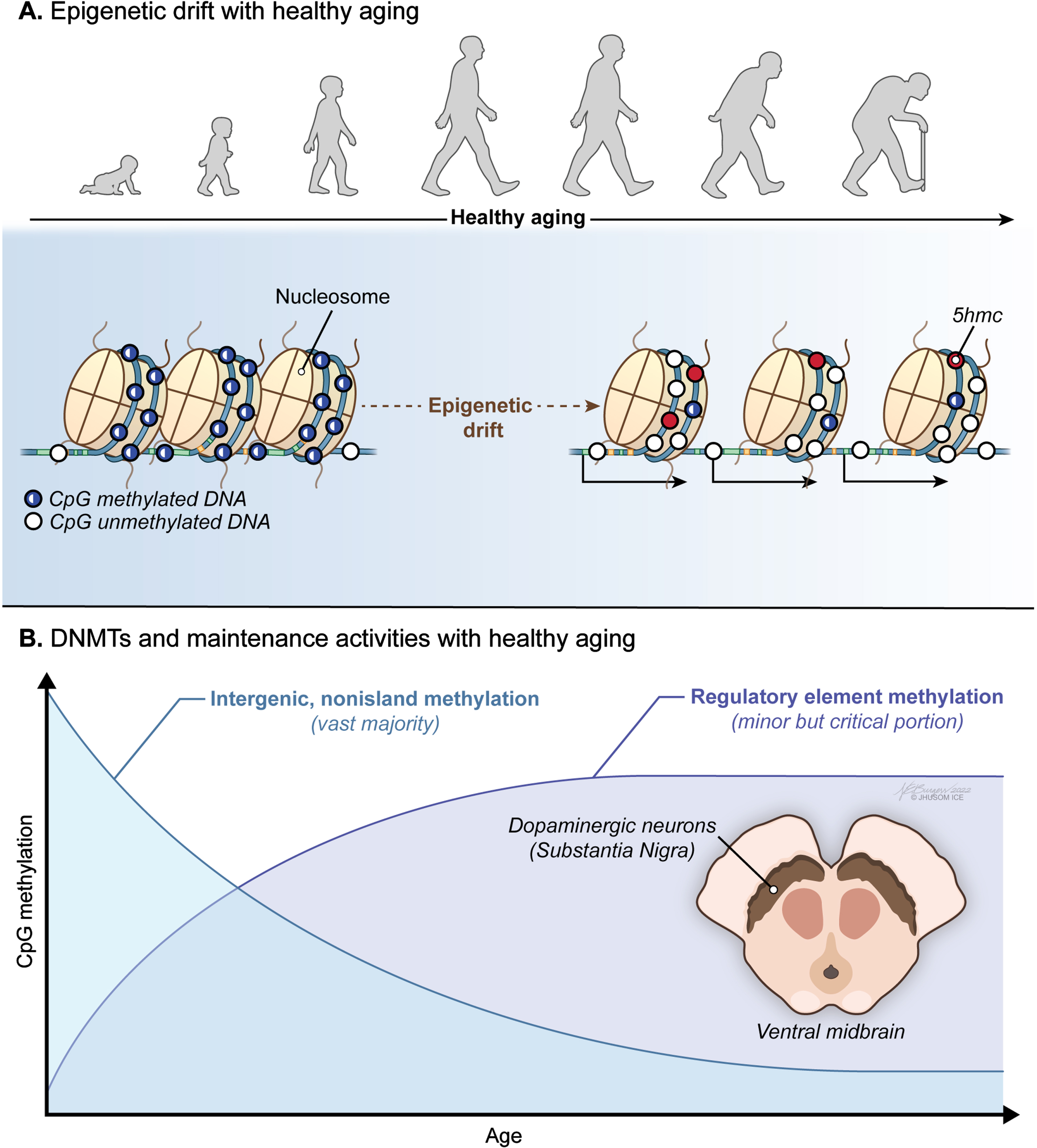
Age-associated trends in DNA methylome of the brain. A) A progressive loss of original epigenetic configurations, also called “epigenetic drift”, is observed during healthy aging. B) A global pattern of DNA hypomethylation with many promoters undergoing aberrant hypermethylation appears to be a hallmark of the aging brain independent of the Parkinson’s disease (PD)-associated neurodegeneration.
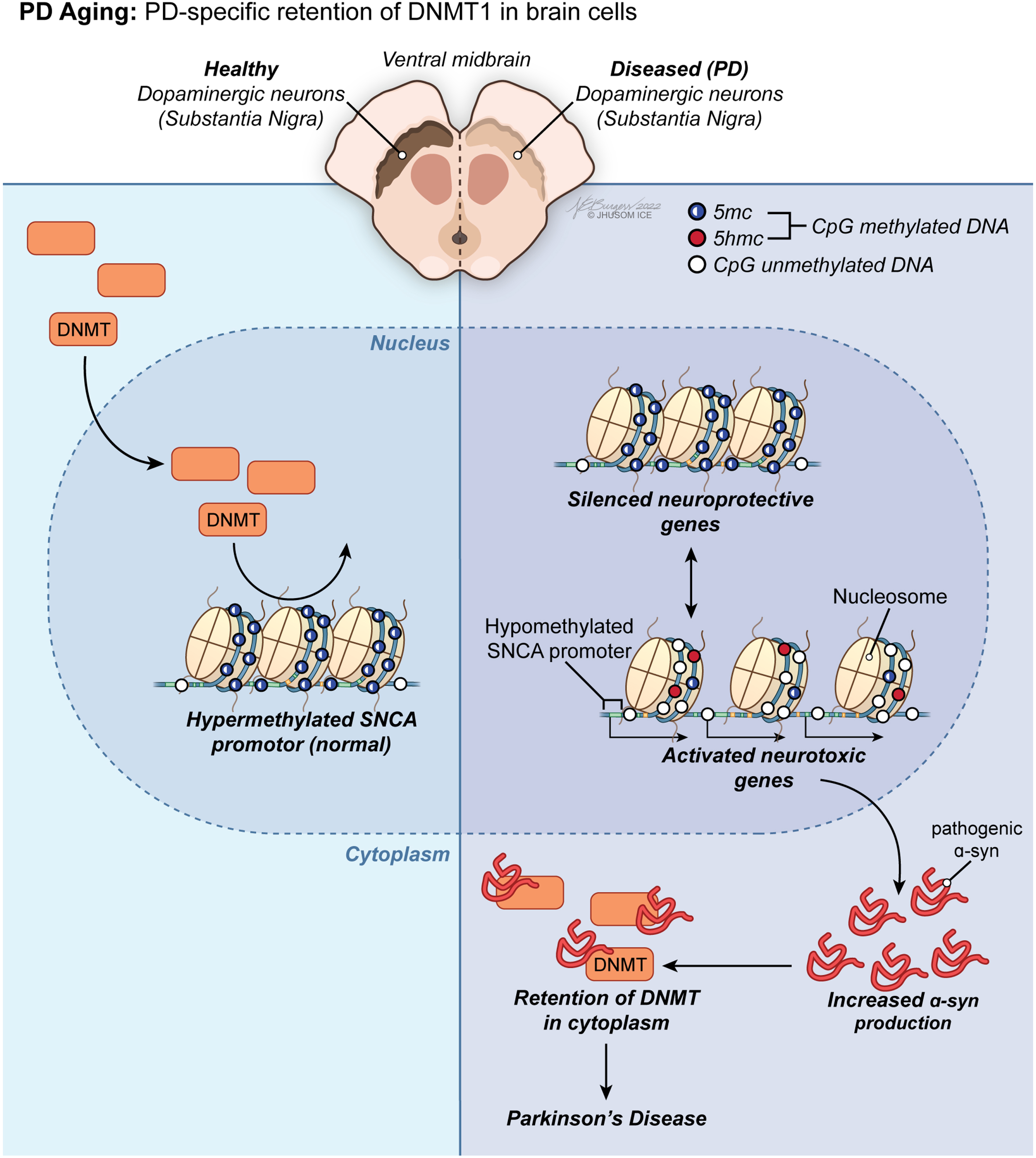
PD aging-associated trends in DNA methylome of the brain. Led by PD-specific retention of DNMT1 in brain cells, a group of methylation anomalies in CpG sites have been linked to aberrant gene silencing or reactivation, including downregulation of neuroprotective genes and overexpression of neurotoxic genes, in PD.
Several factors may account for such conflicting results. Discrepancies between studies might stem from different methylation detection methods with a wide range of sensitivity. Also, variations in cell type composition across different brain regions might yield inconsistent results from one study to another. Lastly, lack of congruity might originate from methodological differences because some use techniques detecting all methylated cytosines whereas others consider (a group of) CpG sites or CGIs [94]. Irrespectively, it remains to be solved as to whether DNA methylome in mammalian brain has an upward or downward slope with age and may indeed be tissue-specific [5, 113]. This applies to site-specific CpGs that show age-associated alterations in methylation pattern, as well. Whereas most sites get hypermethylated with age, some get hypomethylated in an age-dependent manner [26, 114]. Those CpG sites with methylation changes used for predicting the “DNA methylation age” of an organism that correlate well with chronological age are outside the scope of this review and were discussed elsewhere [5].
Dopaminergic neurons in healthy aging
Neuronal cells do not experience replicative aging in mammalian systems, and without degenerative conditions their lifespan depends on the lifespan of the host. In fact, rat neurons were reported to have an increased lifespan up to 100% when transplanted in a longer-living host [115]. Nevertheless, certain regions of the brain and particular types of neurons are more susceptible to degeneration and death associated with the normal course of aging. In this respect, one of these vulnerable cell types reside in the substantia nigra (SN) pars compacta of the midbrain, which contains the cell bodies of dopaminergic neurons contributing to the nigrostriatal tract. During normal aging, these dopamine factories deteriorate with a rate of 5–8% loss per decade of life and demonstrate more pathological changes with healthy aging than any other region of human brain due presumably to excessive production of ROS (highly reactive molecules with unpaired electrons and damaging to biological systems) linked to dopamine metabolism and high energy expenditure of DA neurons [116–118]. And this situation aggravates in the presence of PD-related neurodegeneration, which selectively targets DA neurons. Though the negative correlation between aging (and PD) and the survival of DA neurons has long been acknowledged, the molecular mechanisms underlying this elevated vulnerability of DA neurons to cell loss associated with age is yet to be solved. Given the association of the dynamic nature of DNA (de)methylation with the pathophysiology of aging and PD, research investigating SN-specific signature of healthy aging in the DNA methylome is critical in understanding the breadth and the extent of age-dependent epigenetic contributions in PD.
Changes in the DNA methylome of aging substantia nigra
There is still limited number of studies that assessed age-related alterations in CpG methylation of the SN no matter how tempting it is to go further into the hitherto uncharted territories of the brain. Few evidence over the past few years toward global hypomethylation in the SN with aging has accumulated in the literature [7, 46], though without any solid conclusions. Key findings include: 1) there are global changes in DNA methylome of the SN with age, 2) the ratio of two major forms of epigenetic DNA modifications “5hmC/5mC” in brain significantly increases in the SN with age, implying more DNA demethylation by TET enzymes, 3) there is a tissue-specific increase in 5hmC with age in the DA neurons of the SN, 4) the activity but not the mRNA level of TET enzymes increases in the SN of the aged mice. Yet, none of these DA neuron-specific observations provide direct evidence for an age-dependent change in total 5mC. This issue was addressed within the scope of the same studies, but no significant alteration could be noted. Also, no significant changes in expression or methylation levels of both DNMTs, DNMT1 and DNMT3a, with age were reported in SN, unlike some other regions of the brain discussed above. Independent of all these scientific endeavors, there is also little to no indication of promoter-level changes in DNA methylome of the SN associated with the normal course of aging. Collectively, though the preliminary findings herein lay the groundwork for further examination of alterations in DNA methylome of the SN observed during healthy aging, there is an urgent need for additional research to both uncover the extent to which DNA methylation changes with aging in the SN and examine the reproducibility and generalizability of these findings.
PD and aging: Different sides of the same coin, or two different coins?
Aging appears as the greatest risk factor for sporadic PD, which accounts for a vast majority of PD cases [118]. In other words, a currently unknown set of age-dependent factors predispose a group of individuals to the most common type of PD. More specifically, PD prevalence changes from low levels in young age groups to 15% for individuals over 60 years of age and rises another 15% in every following decade, making up 1% overall prevalence over age 60 [119]. Yet, how advancing age accelerates degeneration of dopaminergic neurons in the SN and thereby substantially increases the likelihood of developing PD is still an open question. As indicated before, genome-wide changes in DNA methylomes observed in PD and in healthy aging of similar age bear resemblance to some extent. In general terms, aging diminishes the overall DNA methylation in the genome and selectively increases levels of methylation at many promoters, roughly like PD. This resemblance of changes in DNA methylome patterns might mean age-dependent epigenetic alterations potentially contribute to the onset and/or progression of PD, which is an age-related disorder [114]. However, this claim demands further evidence that help quantify the degree of resemblance in a tissue-specific manner. From a technical standpoint, more longitudinal studies of large cohorts dissecting the kinetics and dynamics of DNA methylation changes in PD and in healthy aging using time-series data, which might require the use of such peripheral surrogate tissues as blood, are needed to define the sequence of causal events occurring from birth and to measure the degree of correlation between these two states of health. Ultimately, this will help understand the biological underpinnings and pathophysiological characteristics of PD within the context of aging.
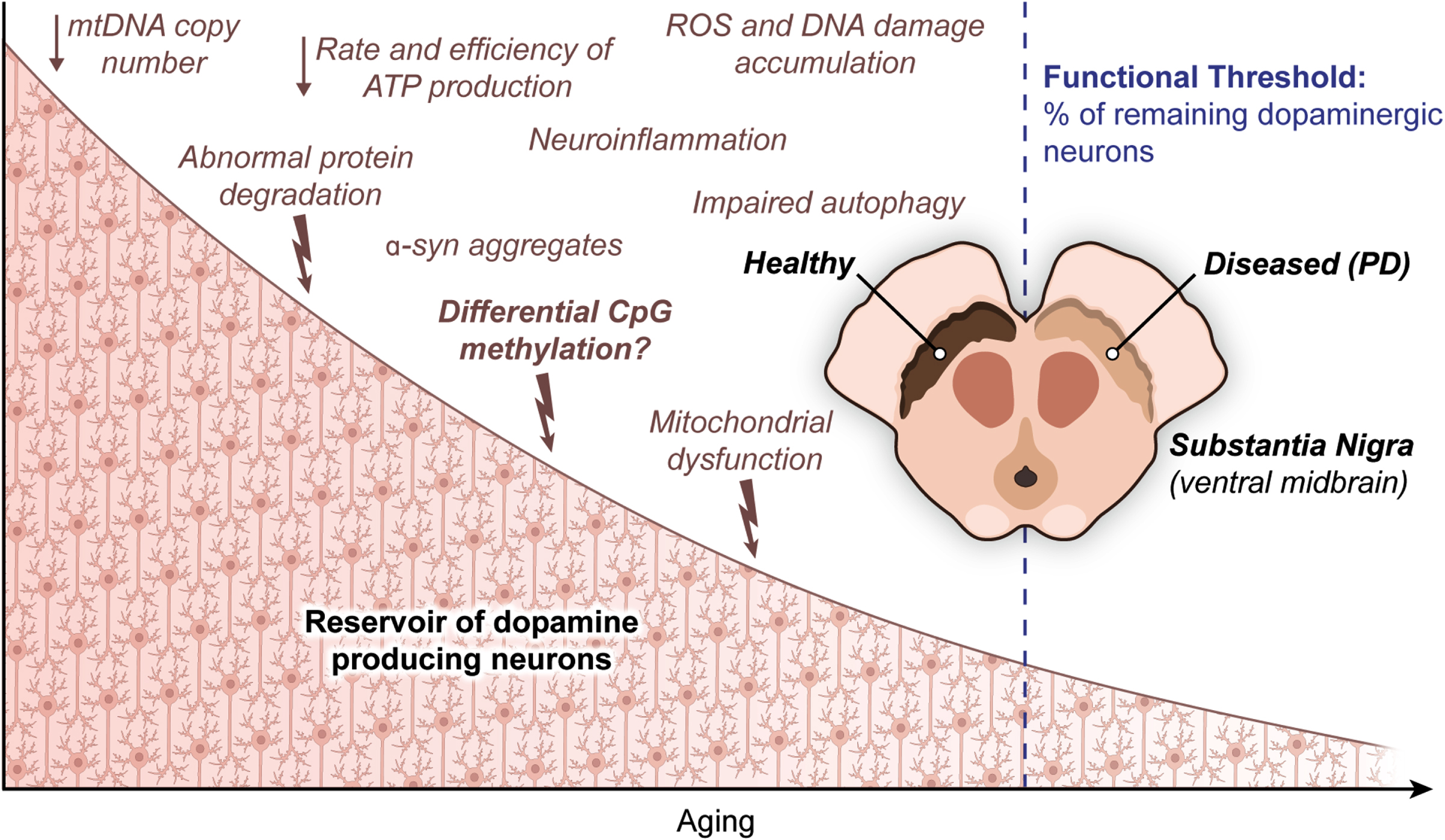
From a biological perspective, a cascade of stressors that arise as a part of the normal course of aging in the SN take a toll on the ability of DA neurons to tolerate further insults as PD progresses. More specifically, advancing age, similar to PD [120], brings along a decline of mitochondrial DNA copy number (mtDNA-CN) and defects in protein degradation together with an increasing burden of dopamine metabolism [118]. While dopamine metabolism likely contributes to neurodegeneration in the SN by the overaccumulation of ROS and subsequent DNA damage over time [121], an ongoing reduction in mtDNA-CN brings about a drop in both rate and efficiency of ATP production required to meet high energy demand of DA neurons [122]. At the same time, abnormal protein degradation adds to neurodegeneration by severely interrupting proper functioning of dopaminergic neurons, as can be seen in PD caused by α-syn aggregates [123]. There is further evidence on both impaired intracellular clearance mechanisms and cytoplasmic lipofuscin mass (a direct consequence of lysosomal reactions related to a major cellular recycling system called autophagy) in DA neurons of rhesus monkey brain [124]. Evidence from nonhuman primates showed an age-dependent decrease in tyrosine hydroxylase (TH), the gold standard marker for dopaminergic neurons, in the SN [125]. Yet another study demonstrated that genetically interfering with aging in the worm C. elegans leads to a substantial increase in life expectancy with significantly less dopaminergic neuron death and clear improvements in behavior deficits associated with these neurons [126]. It has also been shown that inducing aging in iPSCs results in progressive loss of tyrosine hydroxylase, a sign of neurodegeneration in the SN [127]. Finally, the annual killifish N. furzeri, which was found to exhibit an age-dependent degeneration of DA neurons as a part of the natural life cycle and to develop inclusion bodies with α-synuclein in the brain, was now tested as a likely animal model to investigate mechanistic underpinnings of Parkinson’s disease, with aging being part of the equation [128]. From a mechanistic point of view, neuromelanin accumulation progressively increasing with age [129] and the diminishing ability of mitochondria in DA neurons to handle intracellular levels of calcium and/or iron and the following deficiencies in mitochondrial complex I and IV activity [130, 131] all seem epiphenomena of an underlying aging process in the context of PD. On top of this naturally occurring cellular degeneration, the accumulation of random mutations during later stages of development of matured neurons may become a final burden to the already overwhelmed neuronal cells in the SN of the elderly [132]. In this context, heterozygous SNPs in CpG dinucleotides were also found to disrupt the methylation potential of the corresponding sequences [20]. Please see [20] for an excellent work discussing this genetically driven epigenetic changes (that is, allele-specific methylation) in the human genome and [133] for an exquisite review of the impact on healthy aging and PD of the environmental factors/exposures that are known to influence CpG methylation patterns. Briefly, cumulative exposure to risk factors, such as a number of pesticides, has been suggested to contribute to PD more significantly in old age while certain lifestyle behaviors, including but not limited to tobacco smoking and lifelong physical exercise, have been defined as the protective factors for parkinsonism [134]. Overall, genetics and environmental factors cover just a small percentage of PD cases individually. An interesting hypothesis claiming reprogramming to recover youthful epigenetic information might reverse some of the physiological trends observed as a part of healthy aging process in murine system has recently been proposed, though met with some controversy [27].
A key point in PD research is that PD symptoms start to appear only after the total loss of DA neurons reaches a certain level. Including aberrant DNA methylation, anything which increases the degree of vulnerability to accumulating damage in the SN might shed light on why only a fraction, not all, of the elderly population experiences excessive loss of dopaminergic neurons. Given that the regulation of genomic methylation is important for healthy cognitive functions and that the temporal regulation of de novo (de)methylation contributes substantially to the differentiation and maturation of the cerebrospinal axis in mammals [135], CpG methylation can as well be the missing piece of the puzzle that will bridge the persisting gap between health and disease. Together, a range of age-related naturally accumulating stresses derived from different cell compartments undermine cellular tolerance of DA neurons to additional insults of mitochondrial dysfunction and toxic protein aggregates, rendering neurons even more vulnerable to degeneration.
Gender-specific changes in DNA methylomes of PD and aging
One last point is that age-related DNA methylation changes that are potentially sex-specific have been suggested to associate with differential susceptibility to PD in human [136], though without direct experimental evidence. In this respect, our current knowledge includes: 1) females have a higher life expectancy than males [137], 2) the epigenetic aging rate of males is higher than females [24], 3) genes with CpG sites predictive of chronological age in the context of epigenetic aging were found highly associated with age-related neurodegeneration as a potential example of the interplay between aging, DNA methylation, and PD [24, 26], 4) males seem significantly more likely to develop PD than females [138–140]. This potential differential predisposition to PD outcome between males and females might be explained by the changes in autosomal CpG methylation at particular sites observed between different sexes with aging considering both reliable predictions of chronological age with high accuracy using age-dependent methylation changes in just a set of specific CpG sites [5, 24] and the high degree of association of nearby genes with age-related neurodegeneration. Another line of evidence, though indirectly, came from a large cohort study showing that methylation pattern of gender-specific genes, such as MAPT, is positively associated with aging and that gender was a significant independent predictor of this observed methylation pattern [83]. Given the statistical nature of the analysis, further investigation is needed to validate these findings experimentally and to rule out both any spurious correlations and any possibility of confounding variables that are not accounted for in these studies.
Generating methylation data for age-related human diseases
In this respect, high-dimensional data from multiple models should be generated and combined before reporting the findings intended to characterize the genetic-epigenetic pathophysiology of aging in a tissue-specific manner because none of the models alone can fully phenocopy complex human diseases [141]. In general terms, the complex nature of aging at the tissue or organism level can be further illustrated by the common observation that not all aging individuals share the same set of aging phenotypes. From an epigenetics standpoint, a high degree of interindividual variation in the severity and spectrum of aging phenotypes can be largely attributed to a greater level of genetic heterogeneity and a wide range of environmental exposures. In addition to choosing the appropriate ensemble of transgenic animal models to study SN-specific CpG methylation changes with age, the choice of detection method to identify and quantify genome-wide DNA methylation has a tremendous impact on the outcome. Though unable to distinguish between 5mC and 5hmC, whole genome bisulfite sequencing (WGBS) is the gold standard method in DNA methylation studies among a plethora of currently available techniques [142]. Some of the inherent issues related to WGBS coupled with Fluorescence-activated cell sorting include technical challenges related to generation and subsequent use of bisulfite-converted DNA, severe genomic fragmentation during sample preparation and subsequent biases in sequencing data, and difficulties in amplifying long DNA regions [28]. There are also other methods used to detect DNA methylation in a cell type-specific fashion when combined with auxiliary techniques, including but not limited to antibody-based strategies or oxidative bisulfite sequencing, highly sensitive LC–MS/MS and LC–MS/MS/MS, and single cell approaches [143]. Each method has its own merits and drawbacks, and each is at a different phase of development. Please see [142] for an exquisite review of the current methods within the scope of DNA methylation analysis.
Parkinson’s disease models: A platform for cell type-specific DNA methylation studies
Considering that the brains of living subjects are inaccessible, the neuroscience research faces a dilemma of cause and effect due to the absence of empirically robust, real-time evidence from the primarily affected tissue. A substantial number of research focusing on epigenetics of neurodegeneration are based heavily on postmortem brain tissue, which makes it rather difficult to figure out as to whether observed changes in DNA methylome are caused by or leads to neurodegenerative processes. In other words, reprogramming DNA methylation patterns is a major means of propagating the impact caused by environmental exposures on the host at the genome level. At the same time, any change in DNA methylation might as well be a direct or indirect consequence of the sequence of molecular events that lead to the phenotype itself. This, in turn, leads to a causal conundrum in the field. The difficulties in selecting participants and in acquiring/preparing well-characterized high quality specimens at large quantities, the low number of remaining DA neurons due to the end-stage of the disease, potential confounds (e.g., chronic treatments and the presence of comorbid conditions) all add to this picture, as well [144, 145]. Therefore, after an increasing body of evidence suggest that similar alterations occur in both brain and blood methylome of PD patients, many studies were designed to use more accessible systemic tissues [84, 146]. Although there are a few concerns, in addition to the inherent issues related to disease biomarkers, about to what extent epigenetic changes identified in systemic tissues reflect neurodegeneration observed in the brain [6], human blood might serve as a feasible surrogate for brain tissue depending on the study design. It offers such unique advantages as convenient accessibility anytime, the ability to reach higher sample sizes, suitability for high-throughput assays, and relatively high degree of cross-tissue correlation, reflecting host-derived genetic architecture [147–149]. In fact, the view of what PD is has recently shifted away from it simply being considered as a disease of DA neurons to a more complex disease that involves, in addition to brain, many other non-brain regions as well, including the peripheral immune system and possibly the gut and peripheral nervous system (which are rather relevant to the etiology of PD and to prodromal stages prior to diagnosis) [150]. Given that DNA methylation acts differently on gene regulation in different tissues and that epigenetic regulation might indeed be a highly cell type-specific molecular event, one might still think studying tissues that are not primarily affected by the PD-induced neurotoxicity does not seem to have much use in this context. Yet, depending on the nature of the research and the purpose of the study, human blood might indeed be an ideal choice of platform to investigate DNA methylation in neurodegenerative disorders, including PD. In general terms, though, human postmortem studies will likely continue to lay the foundation of our understanding of PD for some more time [145].
Another approach includes the use of an ever-expanding repertoire of human cellular models of neurodegeneration, in particular, induced pluripotent stem cell (iPSC)-derived human neurons [151]. This model makes it possible to explore individual-specific neuronal mechanisms reflecting the unique genetic makeup of each patient [147]. Pluripotency (i.e., the ability to reproduce all adult cell types, including many types of cells in the central nervous system, along the differentiation process), the ability to model disease, and accessibility to large number of patients without any ethical concerns are also part of the package [152]. On the other hand, in addition to labor-, time-, and money-intensive nature of this approach, these models are as well limited regarding the degree of maturation, the efficiency of reprogramming, and the level of complexity in the absence of: 1) complex neuronal circuits, 2) glial cell ramification at full scale, 3) immunovascular components [153]. Though iPSCs offer an ideal platform to generate DA neurons from the patient sample that retain parts of epigenetic signatures of the tissue of origin [154], all these signify the importance of developing engineered animal models that can more faithfully recapitulate the complexity of an intact nervous system as well as the protracted, age-dependent progression and selective neurodegeneration of PD. Yet, it is hitherto widely known that no animal model alone could do so fully, with important PD hallmarks missing in the phenotype [124, 153]. To illustrate, a known issue with the current animal models is as follows: models of α-synuclein-induced degeneration that transgenically express either wild type or various mutant forms of α-synuclein develop the aggregates, yet mostly without the SN-specific neurodegeneration seen in human PD [153]. Also, pharmacological and toxin-induced animal models depended typically on single doses of large amounts of neurotoxic agents fail to develop the common (i.e., not early onset, gradually progressive) deterioration observed in patients with Parkinson’s disease [124]. There are also other transgenic animal models used in PD research, including but not limited to models of LRRK2, PARK2 and PINK1 mutants, each with a different set of challenges. For a detailed overview of the current animal models of neurodegenerative diseases, please see [153]. Together, it is commonly appreciated in the field that not a single, but a diligently selected group of existing models collectively demonstrate key features of PD. Therefore, future studies using this combinatorial approach are pivotal to our understanding of the pathophysiology of PD and will pave the way for more extensive epigenetic research investigating PD-specific changes in DNA methylome of the SN.
FUTURE PERSPECTIVES
Accumulating evidence has revealed that regulating the regulators of DNA methylation, such as DNMTs and TETs, is a key aspect of CpG methylation-based modulation of gene expression within the context of aging and PD [155–159]. More specifically, unbalanced PARylation has been identified as a leading factor contributing to aberrant genomic methylation, as well as changes in chromatin landscape linked to defective genome structure [160]. PARylation is, in general terms, associated with preservation of unmethylated state of CGIs and the genome overall [161], despite some evidence on its functional divergence based on local chromatin topology. The enzyme that catalyzes PARylation reactions, PARP1, controls CpG methylation by suppressing DNMT1 activity both directly and indirectly [160, 162], and by forming a complex with CTCF, a multifunctional structural protein that maintains the stability of local methylation status of many highly expressed genes, to ensure transcriptional activity [163, 164] (Fig. 4). Orchestrating multiple epigenetic mechanisms, PARP1 also controls CpG demethylation by inducing TET1 expression and stimulating its activity, as shown in a group of studies [165, 166]. For mechanistic insights into this post-translational modification in the context of epigenetics, please see [161].
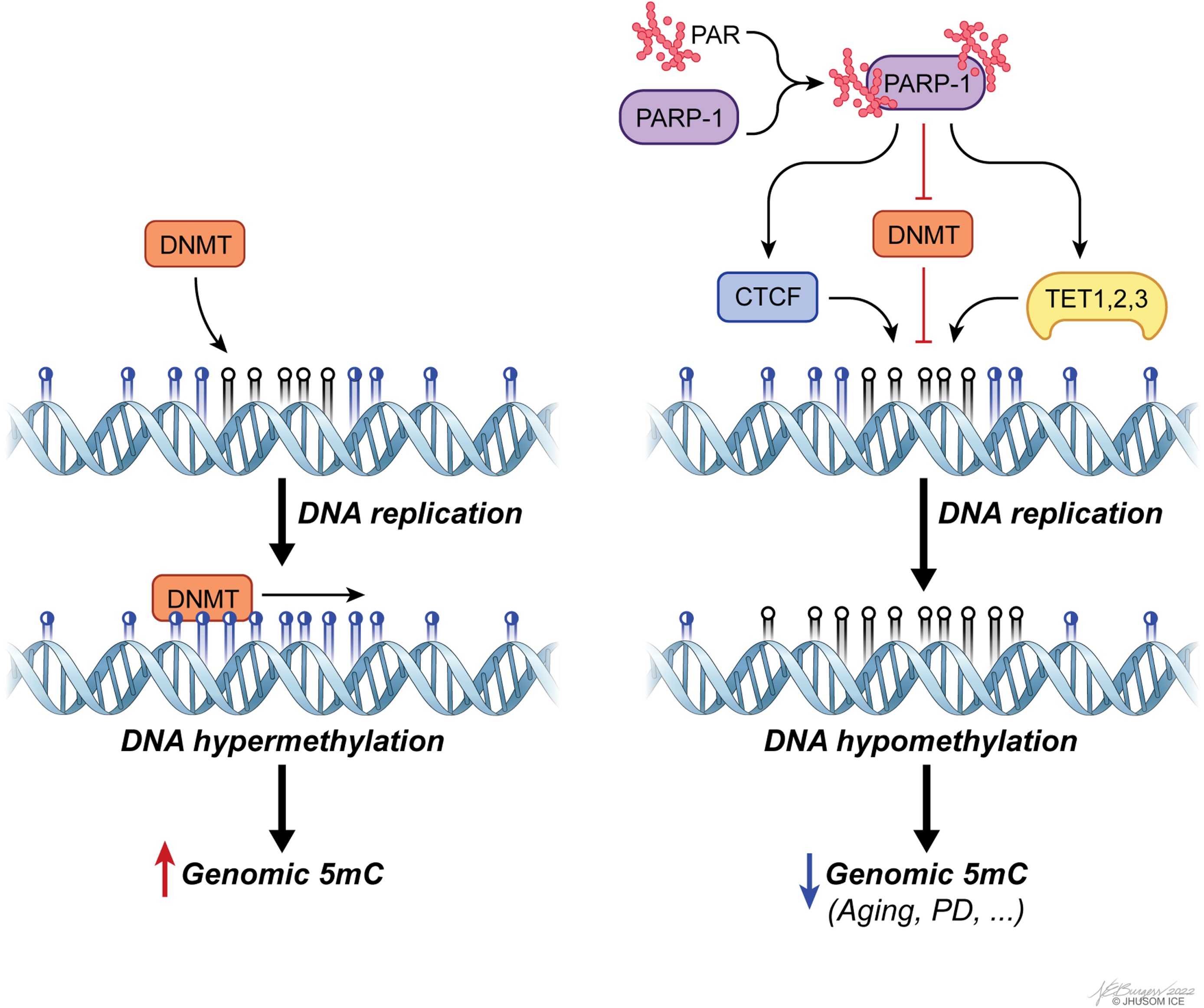
poly(ADP-ribosyl)ation (PARylation) of PAR polymerase 1 (PARP-1) is a major means of regulating genomic methylation patterns. PARylated PARP-1 acts on a set of epigenetics regulators, including CTCF, DNMTs, and TETs, to induce DNA hypomethylation.
There is now strong experimental evidence that PAR-dependent cell death, or parthanatos, in neurons following the accumulation of PAR by overactive PARP1 is involved in both PD [159] and many aspects of aging [167]. In addition to the converging trajectories within the scope of parthanatos of parkinsonism and aging, the well-founded correlation of DNA methylation defects with these two states of health discussed above raises new biological questions: To what extent in DNA methylome of the SN do age-dependent changes correlate with PD-specific alterations? Can PD-specific alterations in DNA methylation independent of aging explain the reason why only some, not all, individuals develop PD with advancing age? Can new animal models of PD that take aging into account phenocopy the multifactorial underpinnings of PD-associated neurodegeneration better than the current animal models do? Can repurposing of PARP-inhibitor drugs currently available on the market be useful for the treatment of PD? In the same context, can the epigenetic burden of aging actually be reversed, as suggested in [27]? The last question, in particular, deserves attention. It has been empirically shown that mammals, including human, maintain a copy of juvenile epigenetic profile at the tissue level and that reversing the epigenetic clock in aged cells through TET1/2-dependent epigenetic reprogramming can successfully recapture youthful gene function, restoring a degenerative phenotype associated with aging in old mice [27]. Though not reproduced in the literature, these observations might have important implications in the functional reversal of age-related alterations in DNA methylome and might even pave the way for the reversal of age-related decline in humans one day. Given the phenomenon that exceptionally long-lived individuals have a slower aging rate (or likely a healthy longevity) calculated using the epigenetic clock [168], critical questions that remain include: will it ever be possible to achieve the same reversal for PD, as well? Can the onset and/or progression of PD be delayed without the age-specific epigenetic component contributing to the disease pathogenesis? Further research is needed to test the validity of these questions.
CONCLUDING REMARKS
From an evolutionary standpoint, the potential to adapt to changing environmental conditions contributes to the fitness of an organism. At the molecular scale, this translates into the following: any additional layer of transcriptome regulations that provides stability and mediates adaptive changes of transcriptional programs greatly helps an organism in adapting to its surrounding. Epigenetic mechanisms are one of the major means of regulating the mammalian transcriptome. Of a whole spectrum of epigenetic processes, DNA methylation is among the best-defined epigenetic processes in neuroscience. We now know that aberrant CpG methylation is one of the factors associated with aging process and age-related neurodegenerative disorders, more specifically PD. In this regard, the dynamic nature of repressive CpG methylation has provided a fertile ground for basic research in these fields over the last few decades. Yet, it is still not known as to whether epigenetic dysregulation is just an epiphenomenon rather than a primary cause of both PD and age-associated decline overall.
Being an evolutionarily conserved natural process, healthy aging is typically defined as subclinical SN neuronal decline caused typically by elevated oxidative stress, mitochondrial dysfunction, derailed autophagy, intracellular α-synuclein aggregates and neuroinflammation, many of which are in line with the pathophysiology of PD [169]. Also, PD is widely known to be caused by a multifactorial interplay of aging, genes and the environment, with aging being the greatest risk factor of all. Despite all these commonalities, the field is far from reaching a consensus in the absence of an ideal animal model that also takes aging into account to study molecular mechanisms underlying PD. Given the importance of defective DNA methylation in the pathophysiology of aging and PD, future research investigating SN-specific signature of healthy aging in DNA methylome is critical in understanding the breadth and the extent of age-dependent epigenetic contributions in PD. Forthcoming technologies, such as high efficiency single cell DNA methylome profiling and complementary multi-omics approaches, to build integrated molecular networks of PD and aging have the potential to accelerate discoveries in this exciting field.
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation and the Diana Helis Henry Medical Research Foundation through their direct engagement in the continuous active conduct of medical research in conjunction with the Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Program. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The figures for this article were illustrated by Noelle Burgess. This work was supported by the JPB Foundation. Adrienne Helis Malvin Medical Research Foundation, Diana Helis Henry Medical Research Foundation. S.U.K. is currently supported from the NINDS/NIH (1R01NS123456-01).
CONFLICT OF INTEREST
The value of patents owned by Valted, LLC could be affected by the review described in this article. Dr. T. and V. Dawson are founders of Valted, LLC and hold an ownership equity interest in the company. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict-of-interest policies. The authors do not have any conflict of interest to report.