
Abstract
Environmental risk factors and gene-environment interactions play a critical role in Parkinson’s disease (PD). However, the relatively large contribution of environmental risk factors in the overwhelming majority of PD cases has been widely neglected in the field. A “PD prevention agenda” proposed in this journal laid out a set of research priorities focused on preventing PD through modification of environmental risk factors. This agenda includes a call for preclinical studies to employ new high-throughput methods for analyzing transcriptomics and epigenomics to provide a deeper understanding of the effects of exposures linked to PD. Here, we focus on epitranscriptomics as a novel area of research with the potential to add to our understanding of the interplay between genes and environmental exposures in PD. Both epigenetics and epitranscriptomics have been recognized as potential mediators of the complex relationship between genes, environment, and disease. Multiple studies have identified epigenetic alterations, such as DNA methylation, associated with PD and PD-related exposures in human studies and preclinical models. In addition, recent technological advancements have made it possible to study epitranscriptomic RNA modifications, such as RNA N6-methyladenosine (m6A), and a handful of recent studies have begun to explore epitranscriptomics in PD-relevant exposure models. Continued exploration of epitranscriptomic mechanisms in environmentally relevant PD models offers the opportunity to identify biomarkers, pre-degenerative changes that precede symptom onset, and potential mitigation strategies for disease prevention and treatment.
INTRODUCTION
Parkinson’s disease (PD) is the fastest growing neurological disease worldwide and recent estimates of incidence in the US are 1.5 times higher than previous estimates [1–4]. Changing demographics of an aging population and declines in cigarette smoking account for some of this increase; however, the rate of increase outpaces those predicted by these factors alone [1]. In addition, the rate of increase is highest in newly industrialized regions and PD diagnoses within the US cluster in a “PD belt” - regions of the Midwest and Northeast with a history of industrialization [2, 5]. Together, this suggests that environmental factors related to industrialization play a role in PD etiology. Identifying specific factors and elucidating mechanisms by which they increase the risk of PD is critical to reducing the global burden of PD.
While there is debate about the magnitude of the relative contributions of genetic and environmental risk factors in the etiology of sporadic PD (sPD), it is well documented that environmental risk factors and gene-environment interactions play a critical role in disease pathogenesis in most PD cases. Despite this evidence, the role of the environment has been widely neglected in the PD research field [6]. An overwhelming majority of PD research has focused on mutations involved in heritable forms of PD, which account for only 5–10% of PD cases, and genes commonly mutated in the remainder of sPD cases that are not familial or monogenically inherited. Estimates suggest that only about a third of phenotypic variance of sPD can be explained by genetics, further supporting that the environment is a major contributor to PD etiology [7]. These sPD cases are caused by a combination of, and interaction between, genetic and environmental risk factors. Although the focus on disease-linked genes has provided insights into underlying mechanisms of PD pathogenesis, non-genetic factors are understudied and often ignored, hindering the development of comprehensive therapeutic strategies and preventative measures.
In a recent publication in this journal, De Miranda and colleagues proposed an interdisciplinary “PD prevention agenda”—a set of research priorities for both preclinical and clinical research that focuses on preventing PD though modification of environmental factors [6]. As part of this agenda, preclinical studies were encouraged to employ new high-throughput methods for analyzing transcriptomics and epigenomics to provide a deeper understanding of the toxic effects of environmental contaminants linked to PD. Utilizing these methods in environmentally relevant PD models offers the opportunity to identify biomarkers, pre-degenerative changes that precede symptom onset, and potential mitigation strategies for disease prevention and treatment.
Epigenetics, and more recently, the emerging field of epitranscriptomics, have been recognized as potential mediators of the complex relationship between genes, environment and disease because they are sensitive to the environment and regulate gene expression throughout the lifespan (Fig. 1) [8–19]. Thus, exploring both epigenetic and epitranscriptomic changes in PD and models of PD-related exposures is a critical and underexplored avenue of research. By utilizing these high-throughput technologies to explore epigenetic and epitranscriptomic mechanisms in preclinical studies, we can contribute significantly to our understanding of non-genetic factors in PD and to unraveling the complexities of PD etiology.
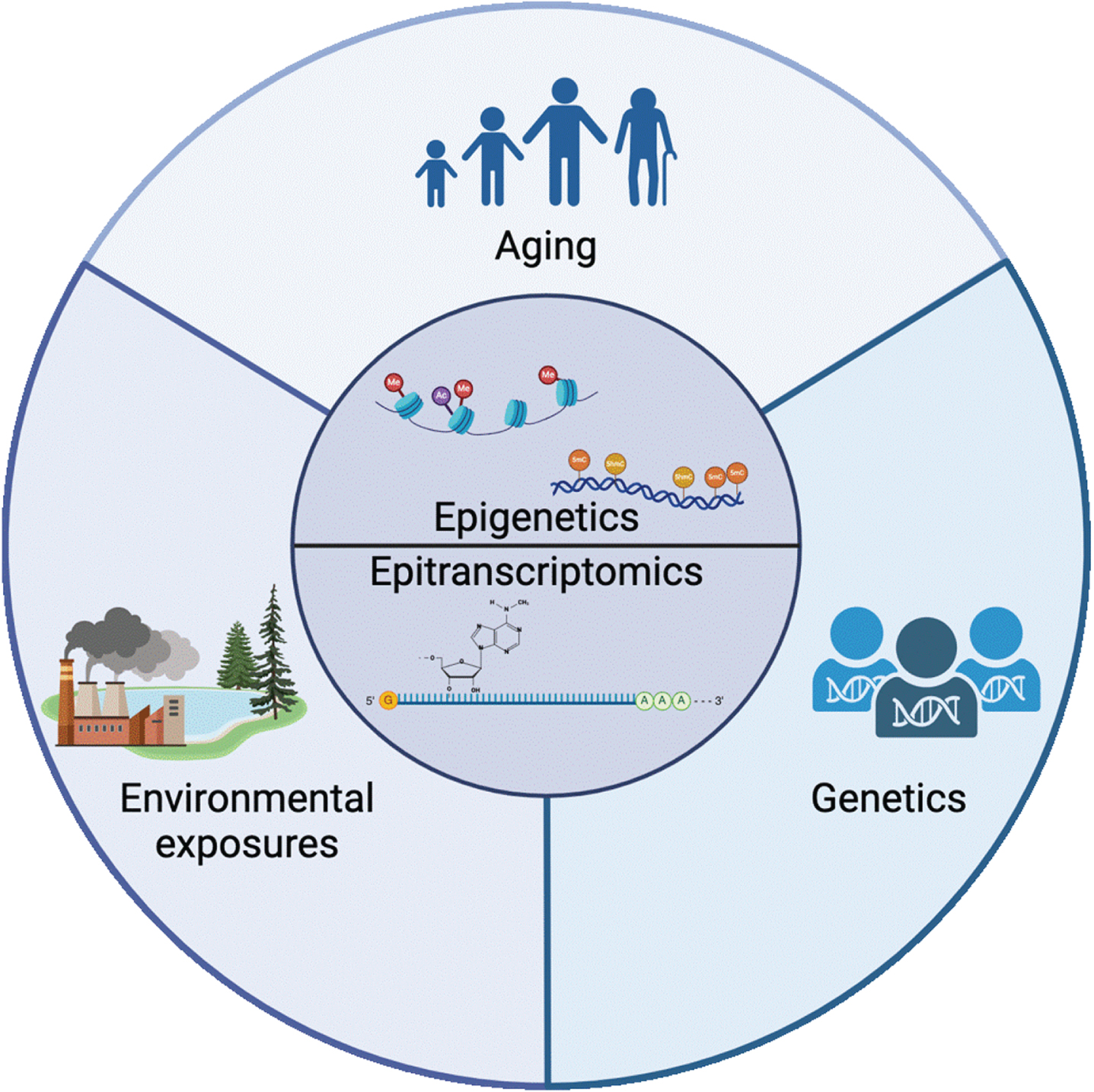
Epigenetics and epitranscriptomics sit at the intersection of the 3 major classes of risk factors for PD: aging, environmental exposure, and genetics. The mechanisms involved in regulating the epigenome and epitranscriptome are mediators of the complex relationship between aging, the environment, and disease. Created with BioRender.com.
EPIGENETICS
Epigenetics is defined as a set of mechanisms that regulate gene expression without modifying the DNA sequence itself and are meiotically and mitotically heritable in dividing cells [20, 21]. It generally includes posttranslational modification of histones and the regulation of chromatin structure, cytosine modifications, and non-coding RNA-mediated mechanisms (Fig. 2) [22–25]. Because epigenetic marks are sensitive to the environment and play a critical role in regulation of gene expression, they are considered a potential mediator of the relationship between genes, the environment, and disease [8–10].
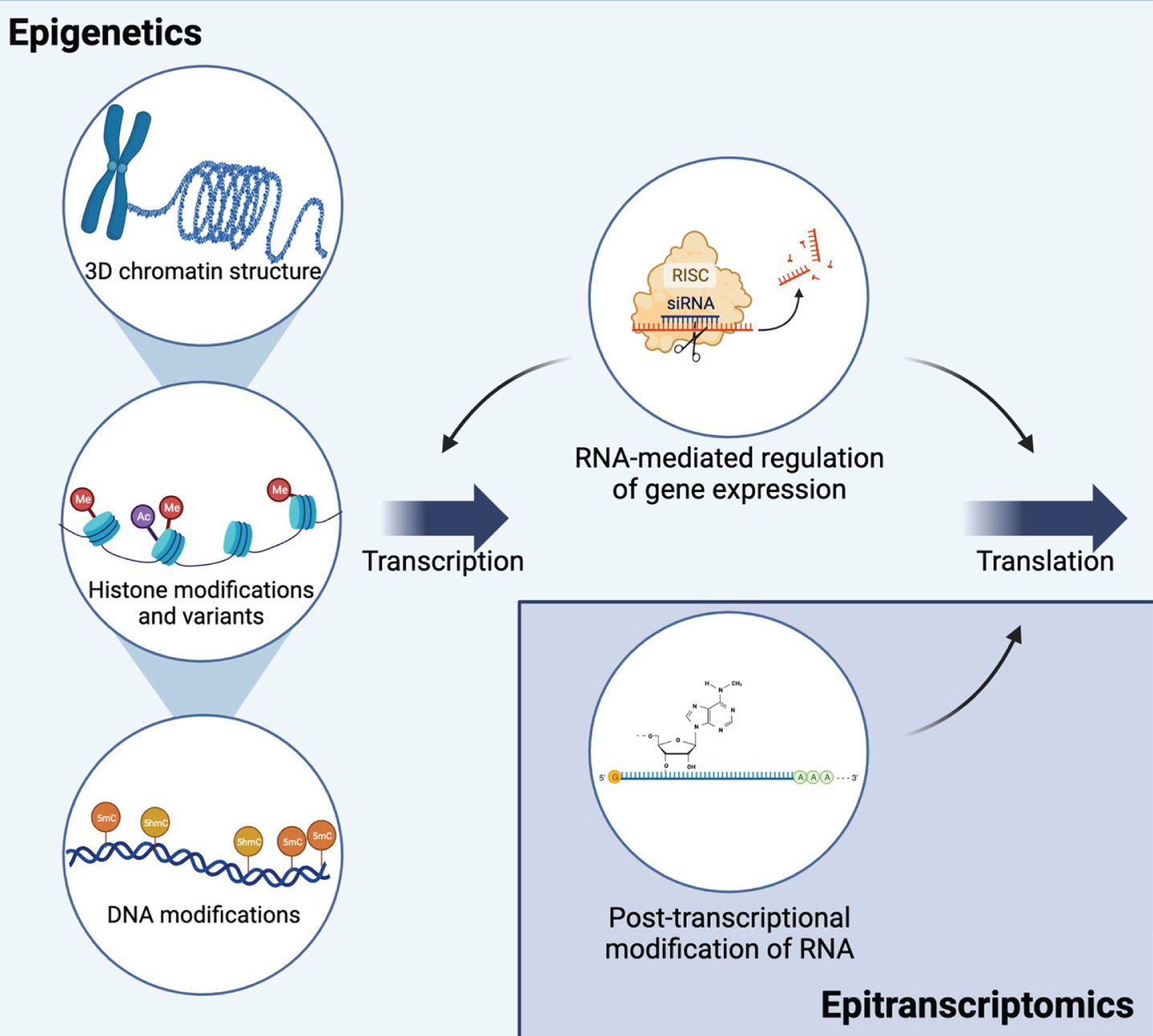
Epigenetic and epitranscriptomic mechanisms interact to regulate gene expression and translation. Epigenetic mechanisms include mechanisms at the DNA level that regulate transcription: 3D chromatin structure, histone modifications and variants, and DNA modifications. Multiple types of non-coding RNAs regulate both transcription and translation. RNA interference is shown as an example of RNA-mediated gene silencing. Epitranscriptomic mechanisms include multiple types of RNA modifications on all types of RNA. The most common mRNA modification, N6-methyladenosine (m6A), is shown as an example. Together, these mechanisms allow for dynamic regulation of transcription and translation and are influenced by environmental exposures. Created with BioRender.com.
One of the most well studied epigenetic marks is the covalent modification of the fifth position of cytosine in DNA (5-methylcytosine, 5 mC) [25]. More recently, further oxidation of 5 mC to 5-hydroxymethylcytosine (5 hmC) has been recognized as a critical epigenetic mark [26–28]. Each of these marks has a distinct set of “writers” and “readers” that catalyze their generation and recognize these marks [26]. Together, these marks play a critical role in the regulation of gene expression. 5 hmC is thought to be particularly important in the central nervous system, where 5 hmC is highly enriched, and the response to neurotoxicants [24, 29].
Epigenetics in PD
Emerging research has shown distinct DNA methylation patterns in individuals with PD compared to controls [23, 30–34]. Specifically, targeted studies have reported differential DNA methylation at multiple PD-related genes including MAPT, CYP2E1, STX1B and the α-syn gene (SNCA) [35–44]. In addition, epigenome-wide analyses of DNA methylation from postmortem PD brain tissue have identified a number of gene regions that show differential DNA methylation in PD brains including genes previously implicated in PD including PARK7, SLC17A6, and NR4A2 and other genes involved in neurodevelopment, neurotransmitter packaging and release, and axon and neuron projection guidance [45–49]. In addition, studies identified an association between a polymorphism in PD risk and the 5 mC “writer”, DNA methyltransferase 3B (DNMT3B), and the 5 hmC “reader”, TET1 [50, 51]. While much less research has been done on histone modifications in PD, a recent study identified hyperacetylation of histone H3K27 in multiple regions of PD brain tissue and altered H3K27 acetylation was found within known PD genes including SNCA, PARK7, PRKN and MAPT [52]. Together, this data suggests that multiple types of epigenetic regulation may play an important role in PD.
Epigenetics in PD-related exposures
The current state of research exploring the relationship between epigenetic mechanisms and environmental risk factors in PD was recently reviewed in a comprehensive systematic review [53]. Overall, studies of PD-related environmental risk factors in animal and cell models support a role for the epigenome, but human studies to date remain limited, largely due to limited access to brain tissue and lack of exposure data for brain bank subjects [11, 54]. Additional studies in this area are critical for continued elucidation of mechanisms of sPD pathogenesis and identification of novel therapeutic strategies. Targeting of genes and proteins that are epigenetically regulated or aberrant epigenetics patterns themselves with precision treatments, such as epigenome-modifying drugs, represent potential new targets and strategies for disease-modifying therapeutics [55]. However, further research is needed to decipher the complexities of these epigenetic changes and their functional implications.
EPITRANSCRIPTOMICS
Epitranscriptomics focuses on post-transcriptional chemical modifications to RNA molecules and their functional impact on translation, as these modifications play key roles in control of multiple levels of RNA metabolism, including stability, splicing, translation, and localization (Fig. 2) [56–58]. RNA modifications have been identified across all forms of life, and in many RNA species, including mRNAs, rRNAs, tRNAs, and small nuclear RNAs [59]. About 170 RNA modifications exist, and the most well-studied include N6-methyladenosine (m6A), inosine (I), pseudouridine (ψ), 5-methylcytosine (m5C), and 5-hydroxymethylcytosine (hm5C) (Fig. 3) [58, 61]. m6A is the most abundant chemical modification of mRNA, and can impact various aspects of RNA function, including RNA splicing, stability, and translation efficiency [58, 62].
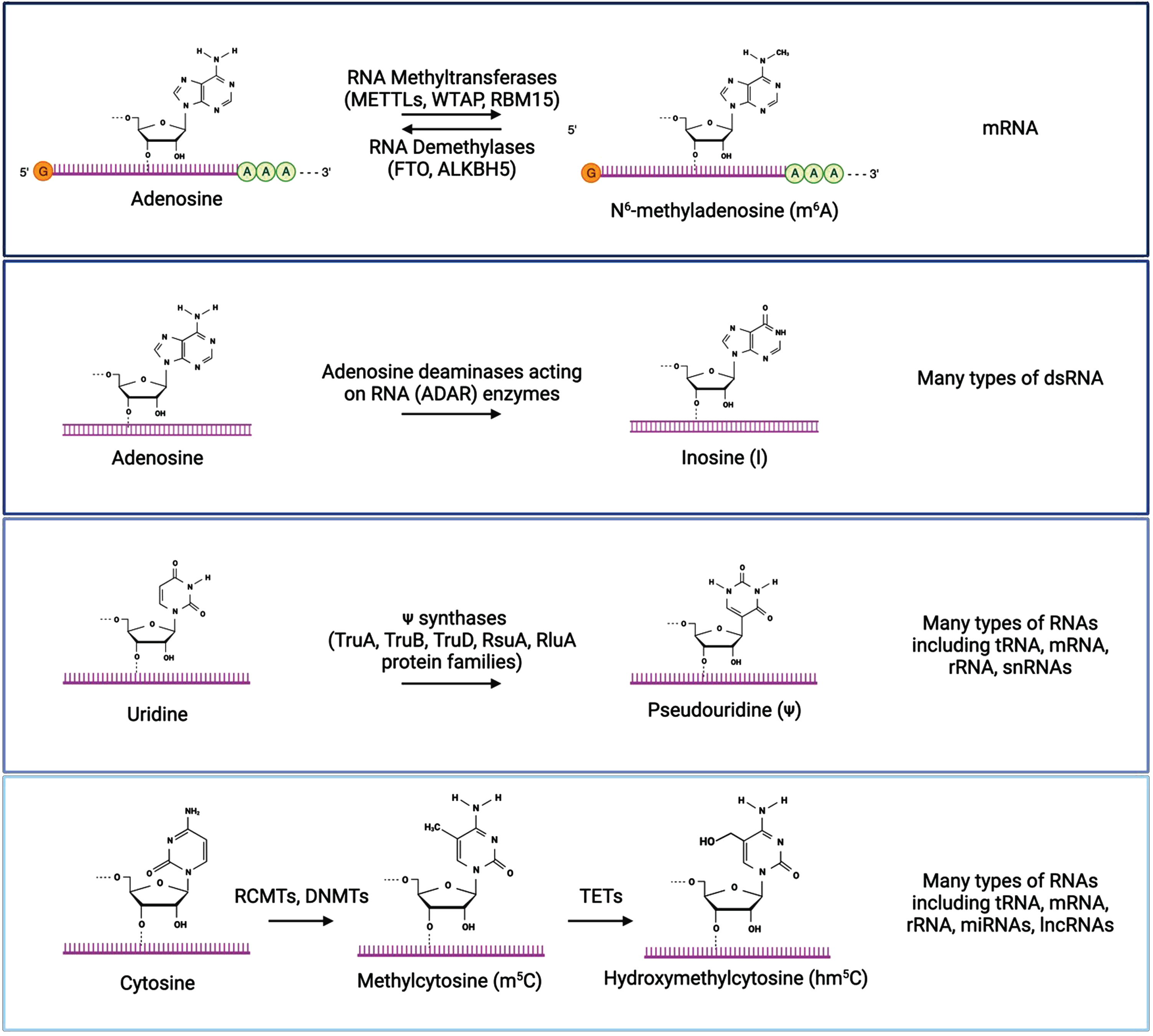
The most well characterized RNA modifications are m6A, inosine, pseudouridine, and cytosine methylation/hydroxymethylation. Chemical structures of modified and unmodified bases are show, along with enzymes that mediate these modifications and the types of RNA molecules where each modification is typically found. Created with BioRender.com.
Inosine is produced by the modification of adenosine to inosine on double-stranded RNA (dsRNA) through the deamination of adenosine by adenosine deaminases acting on RNA (ADAR) enzymes [63–65]. This A-to-I modification is recognized as guanosine by the translational machinery, which impacts translation by altering codons, introducing or removing splice sites, or affecting base pairing of the modified RNA either with itself or other RNAs [66, 67]. Inosine plays in important role in the regulation of RNA editing, alternative splicing, and RNA degradation [63]. Pseudouridine is a modification of uridine installed by pseudouridine synthase enzymes [68–73]. It is the most abundant modified nucleotide on RNAs, and plays a role in RNA structure, splicing, and translation efficiency [69, 70]. Methylcytosine (m5C) is a modification of cytosine in RNA that is converted by RNA (C5-cytosine) methyltransferases (RCMTs) and the same DNMTs that add methyl groups to DNA, [74–77]. Similar to the DNA modification, hydroxymethylcytosine (hm5C) is a further modification of methylcytosine generated by the oxidation of m5C by the TET family of enzymes [78, 79]. These modifications are differentially recognized by reader proteins, which can impact mRNA stability and translation, and these modifications play important roles in the regulation of embryonic stem cell differentiation and neurogenesis [74, 80–83]. Dysregulation of these RNA modifications has been implicated in the development and progression of various diseases, including cancer, neurological diseases, and metabolic disorders [59, 84].
N6-methyladenosine
m6A is the most abundant mRNA modification and plays an important role in RNA stability, mRNA translation, alternative splicing, and subcellular RNA localization [85, 86]. m6A modifications are regulated by complexes of proteins termed “writers”, “erasers”, and “readers.” which have been recently reviewed (Fig. 4) [85, 87]. Writers are methyltransferases that convert adenosine to of m6A using the S-adenosylmethionine (SAM) as the methyl donor to catalyze the formation of a methyl group at the sixth N element of adenine in RNA [88]. The most well-characterized writers include the methyltransferase-like (METTL) family of proteins (METTL3, METTL14, and METTL16), WT1 associated protein (WTAP), RNA binding motif protein 15 (RBM15), and vir-like m6A methyltransferase associated (VIRMA). Erasers are demethylases that assist in m6A removal. There two known proteins in this family –the fat mass and obesity associated (FTO) protein and the AlkB homolog 5 (ALKBH5) protein. Readers are cytosolic or nuclear-localized binding proteins that bind m6A modified RNA and regulate gene expression by altering mRNA stability, splicing, and nuclear export. Readers include the YTH domain-containing proteins (YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3) and heterogeneous nuclear ribonucleoproteins (hnRNPs). These protein complexes carry out dynamic and reversible biological processes that can impact the translatability of RNA and potentially contribute to the pathogenesis of disease states [87, 89–102].

Summary of m6A modulators by class and cellular localization. Created with BioRender.com.
m6A modifications in PD
The importance of RNA modifications, especially m6A, in the development and normal function of the nervous system is well-established and dysregulation of these modifications has been implicated in multiple neurological diseases and aging [85, 103–107]. Studies of m6A in PD models and the human brain have only recently begun to be performed, and more research is needed to develop a comprehensive understanding of m6A activity in relation to PD-associated neurodegeneration.
Existing genetic studies of m6A-SNPs support a role of these modifications in PD. These are SNPs that alter motifs required for m6A modulators to perform their writing, erasing, or reading function or SNPs within the genes encoding these modulators. Two studies that utilized existing GWAS studies, the m6Avar database of m6A-SNPs, existing gene expression data, and eQTL analysis found PD-associated m6A-SNPs within many genes, including the m6A modulator, ALKBH5, which encodes an m6A demethylase; and within motifs required for m6A modulators to function within GAK, which has previously been identified to have two potential PD-linked SNPs in Chinese populations; and ATG16L1, which is important in autophagosome function; and multiple HLA genes [108–111].
A recent analysis of postmortem human PD brain tested for m6A-modified RNAs within neuronal populations in the cerebellum, hippocampus, frontal cortex, and cingulate gyrus [112]. Using a machine learning image analysis method, they concluded m6A-modified RNAs were significantly reduced or mislocalized in cingulate gyrus and hippocampus of subjects with PD, suggesting the possibility of dysregulated epitranscriptomic processes impacting translational control in PD. While this a small preliminary study, this initial finding suggests that further studies of m6A in PD are warranted to confirm and extend these findings.
m6A modifications in PD models
Studies in rodent PD models also support that dysregulation of m6A plays a role in PD (Table 1). A comprehensive study of m6A regulators in the striatum and substantia nigra (SN) of 1-methyl-4-phenyl-1,2,3,6-tetrahydrophyridine (MPTP)-injected C57BL/6 mice (8–12 weeks of age; acute intra-peritoneal injections, 20 mg/kg spread over 8 hours) identified protein expression dysregulation of three m6A writers in the striatum (METTL3, CBLL1, and RBM15), the ALKBH5 eraser in the SN, three readers in the striatum (HNRNPC, IGF2BP3, and FMR1), and four readers in the SN (YTHDF1, HNRNPC, IGF2BP2, and FMR1) by western blot analysis, suggesting dysregulation of the m6A regulatory system in the MPTP model [113]. An additional MPTP study in C57BL/6 mice (4 month old, intra-peritoneal injections, 30 mg/kg/day for 5 days) found increased expression of FTO and decreased expression of METTL14 in the striatum and that exosome-delivery of FTO-targeted siRNAs mitigated the effects of MPTP in the nigrostriatal system [114]. In rats, unilateral SN injection of 6-hydroxydopamine (6-OHDA) (8μg) led to a reduction of m6A and a corresponding elevation of the demethylases FTO in the midbrain and ALKBH5 in the striatum [115]. These findings were recapitulated in PC12 cells where FTO overexpression increased dopaminergic neuronal apoptosis [115]. Collectively, these studies highlight the potential significance of m6A dysregulation in PD pathogenesis and underscore the need to explore these mechanisms in environmentally relevant PD models.
Summary of studies of m6a modulators and modifications in PD-relevant models
Dopaminergic dysfunction in knockout models of m6A modulators
Knockout models of specific regulators of m6A also indicate a role for these proteins within the dopaminergic system. Knockout of the m6A demethylase, FTO, in C57BL/6 mice increases m6A modification within mRNAs important for synaptic transmission and cell-cell signaling in the striatum and midbrain, as assessed by methylated RNA immunoprecipitation coupled with next-generation sequencing (meRIP-seq) and gene ontology (GO) enrichment analysis [116–118]. In both conventional and conditional FTO knockouts, electrophysiological recordings show an impairment of dopamine receptor type 2 (D2R) and type 3 (D3R)-dependent control of neuronal activity and behavioral responses in midbrain dopaminergic neurons [119]. In addition, conditional deletion of the m6A methyltransferase, METTL14, in the SN of C57BL/6 mice (injection of Cre-recombinase lentivirus into SN floxed METTL14 mice at 8–12 weeks) led to a loss of the dopaminergic marker tyrosine hydroxylase in the SN and impaired motor function and locomotor activity assessed by rotarod, pole test, open-field test and elevated plus maze [120]. These knockout models of specific m6A regulators indicate that m6A plays an important role in dopaminergic neuron function and support a need for more focused research in this area. Taken together, a growing body of evidence from human and rodent studies supports an important regulatory role of m6A modifications in dopaminergic neurons.
m6A modifications and environmental exposures
Emerging data from in vitro, rodent, and human exposure studies suggest that, like epigenetic marks, epitranscriptomic marks are also responsive to environmental exposures [12, 15–19]. This rapidly growing body of evidence indicates that a wide range of exposures, including but not limited to polyaromatic hydrocarbons, endocrine disruptors, dioxins, persistent organic pollutants, certain types of pesticides, heavy metals, air pollution, cigarette smoking, and nanoparticles, have been shown to affect RNA modifications and expression of RNA modulators in many types of cells, animal models and human studies. In addition, a small number of studies have explored the effect of environmental exposures on RNA modifications and expression of RNA in brain or in brain-related cell lines. With these new studies, evidence is growing to support that exposure to many types of neurotoxicants (including dioxin, arsenic, lead, atrazine, cobalt, paraquat) affects both expression of RNA modulators and the patterns of RNA modifications in brain and brain-derived cell lines [18, 121–125].
m6A modifications and PD-related environmental exposures
Research in epitranscriptomics in PD-related environmental exposures is in its infancy with only a few studies reported in the literature to date exploring the effects of the pesticide paraquat (PQ) and manganese (Mn) on epitranscriptomic mechanisms (Table 1). Epidemiological studies report an association between exposure to PQ and increased risk of PD, and mechanistic studies in rodents and cell lines support a role for PQ exposure in dopaminergic dysfunction [126–130]. Two recent studies in N2a neuroblastoma cells found that paraquat exposure led to differential m6A modification in circular RNAs and long non-coding RNAs and that these changes in RNA modifications were mediated by oxidative stress [131, 132]. Mn exposure can lead to manganism, an acquired disorder that shares some clinical features with PD, including the motor symptoms of parkinsonism and dopamine dysfunction [133, 134]. One study explored a potential role for RNA modifications in a mouse model of manganese exposure, reporting decreased levels of FTO in striatum after Mn exposure and that overexpression of FTO in the striatum can rescue Mn-induced motor deficits [135]. Despite this small number of studies, evidence is growing to support that RNA modifications are an important mediator of the effects of environmental exposures in many tissues, including brain, underscoring the need for additional research in this area.
CRITICAL CONSIDERATIONS FOR INCORPORATING EPITRANSCRIPTOMICS INTO PRECLINICAL STUDIES OF ENVIRONMENTAL MODELS OF PD
Historically, comprehensive assessments of epigenomic and epitranscriptomic changes faced constraints posed by technology and cost. Development of methods for cost-effect analysis of the epitranscriptome has lagged behind those for the epigenome. However, recent advances allow research to explore these epitranscriptomic mechanisms with heightened precision and cost-effectiveness [49, 137]. The technology is now available to enable the delineation of specific RNA modifications in environmentally relevant PD models. With these new and evolving methods, both genome-wide and targeted analysis of PD-associated genes are possible and warranted to identify pre-degenerative changes and potential mitigation strategies for disease prevention and treatment.
While these new methods provide new opportunities, successful incorporation of epitranscriptomics into PD-relevant exposure models faces challenges, many of which are similar to those faced by the epigenomics and other omics field. In a recently published chapter, entitled “Best practices for epigenome-wide DNA modification data collection and analysis,” we outlined important considerations for rigor and reproducibility in epigenomic studies and recommendations to address them [138]. Many of these challenges and strategies to address them apply to epitranscriptomic studies as well and are summarized in Fig. 5. Most of these practices are not unique to epitranscriptomics and are covered in detail in our previously published chapter [138]. However, the question of the stability of RNA modifications is unique and important to consider the implications of this. In contrast to DNA, many RNA molecules are short lived so modification of individual RNA molecules is also likely short lived and may be difficult to measure consistently. However, as papers discussed above regarding m6A-SNPs demonstrate, if the mechanisms that lay down and regulate these marks are disrupted by toxicant exposure, aggregate changes in RNA modifications may be more persistent that any individual RNA molecule [108–125, 139].
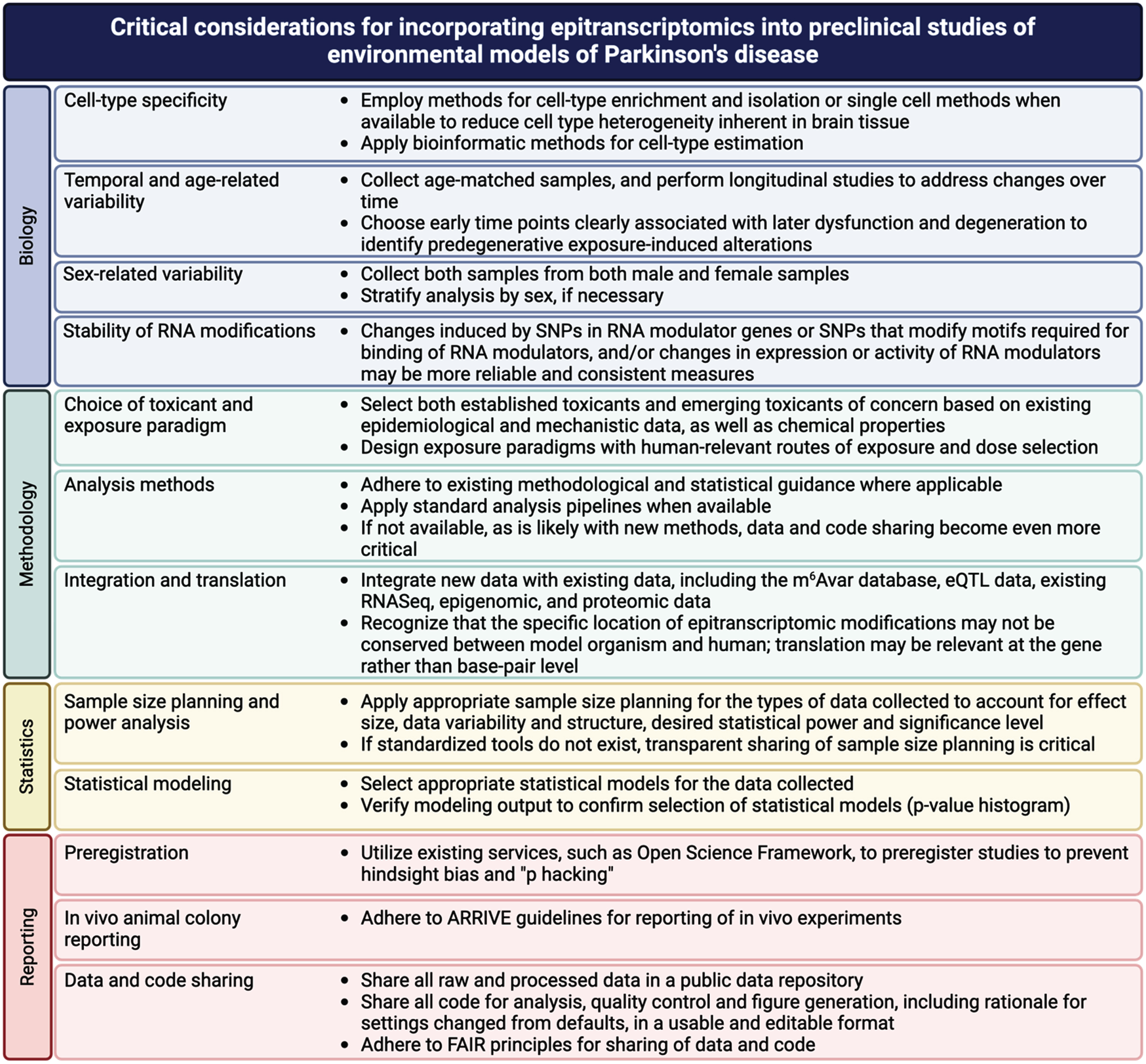
Critical considerations for incorporating epitranscriptomics into preclinical studies of environmental models of Parkinson’s disease.
In addition, like epigenetic marks, the functional implications of epitranscriptomic marks associated with exposure can be difficult to assess. Implementation of the strategies highlighted here regarding integration and translation can help to link these marks with functional outcomes. Much as papers discussed above integrated databases GWAS data and eQTL data with data on RNA modifications (m6Avar), integration of new data will be critical to making functional and translational connections [108–111, 140]. Such integration is only possible with open and FAIR sharing of methods, data and code [141–144]. By building on lessons learned in more established fields, epitranscriptomics can avoid the same pitfalls and facilitate discovery of epitranscriptomic mechanisms in environmentally induced increases in PD risk.
CONCLUSIONS
This review focuses the importance of investigating two interrelated areas of study—epigenetics and epitranscriptomics—in the context of sPD, with a focus on epitranscriptomics as an emerging field of research. While there have been substantial strides in unraveling the basic biology of RNA modifications, the application of these findings to PD remains in its infancy [14, 145]. Given the significant role of environmental factors in sPD and the growing evidence that RNA modifications are sensitive to the environment, further exploration of the role of epitranscriptomics in experimental models of sPD offers a valuable avenue for gaining crucial insights into the molecular underpinnings of sPD.
Combined with the ability to develop targeted therapies that modify epigenetic or epitranscriptomic marks holds substantial promise for mitigation exposure-related risk of PD. The strategic design of small molecules or interventions capable of selectively modulating specific modifications or regulators may provide a nuanced approach to altering gene expression and cellular processes, paving the way for innovative disease-modifying treatments. While much research is still needed to generate a comprehensive understanding of RNA modifications in PD-related exposures and to determine the potential therapeutic and preventative relevance, there is great potential of incorporating new high-throughput methods for analyzing epitranscriptomics and epigenomics to provide a deeper understanding how environmental exposures contribute to sPD and insights into mitigation strategies for disease prevention and treatment. The authors have no acknowledgments to report.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work is supported by NIH R01ES031237.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.